rust-mdbg 一款用于基因组组装的高效率软件
写在前面
rust-mdbg 是一种超快的minimizer-space de Bruijn graphs (mdBG) 实现,适用于组装长而准确的读数,例如PacBio HiFi。
随着18年以来Pacbio HiFi reads的出现,让一些复杂基因组的组装不再复杂,而且有越来越多的课题组也加入到了基因组学的研究中,正是因为有了高精度长读长的reads,目前也产生了很多专门用于HiFi组装的软件,如Hifiasm,当然这篇文章的软件的算法,可以用超短时间,低内存去组装。我相信随着不断的发展,以后做组装的时候甚至都不需要服务器,在个人电脑也可以实现。
内容写了很多,考虑到阅读体验,做了删减。
下面是正文~
原理介绍
DNA测序技术发展的很快,尤其是以Pacbio HiFi数据为代表的Long reads兼顾了长读长以及高准确度。
在这里,研究者定义了一种算法方法mdBG,它利用最小空间德布莱英图(de Bruijn graph)实现long reads基因组组装。
这里插入一个视频,让大家了解一下以前的组装软件用到的 de Bruijn graph的原理
与现有方法相比,mdBG 在速度和内存使用方面都实现了几个数量级的改进,而不会影响准确性。
实战使用8个核心和10 GB RAM在10分钟内组装人类基因组,使用1 GB RAM在4分钟内组装60 GB的宏基因组数据。
此外,研究者构建了661405个细菌基因组的最小空间de Bruijn graph,包括1600万个节点和4500万条边,并在12分钟内成功搜索了抗微生物耐药性(AMR)基因。
鉴于基因组学、宏基因组学和泛基因组学中长读长测序的兴起,预计这一进步对序列分析至关重要!
构建mdBGs的代码可免费下载,网址为
https://github.com/ekimb/rust-mdbg/
介绍
DNA测序数据持续改善,
- 从最开始低质量的长reads,用于组装第一批人类基因组
- 再到Illumina低误差率(%1%)的短reads,
- 目前低误差率的长reads。例如,Pacbio HiFi 1%的错误率产生10-25kbp长(HiFi)reads 进行测序;Nanopore的R10.3孔在~5%的错误率。
未来DNA测序最终将产生长的、近乎完美的reads。
这些新技术要求算法对基因组组装等重要序列分析任务既有效又准确。
在这里,作者为低错误长读长数据提供了一种高效的基因组组装工具(详见后面进展和潜力)。作者引入了最小化空间 de Bruijn 图,mdBGs。
具体来说,每个reads最初都被转换为其最小值(minimizers)的有序序列。minimizers的顺序很重要,因为作者的目标是将整个基因组重建为一个有序列表。
作者的方法不同于经典的MinHash技术,后者将序列转换为无序的最小化集合以检测它们之间的成对相似性。为了帮助组装更高错误率的数据,作者还引入了偏序比对 (POA) 算法的一种变体,该算法在最小化空间(minimizer space)而不是基础空间(base space)中运行,并有效地纠正与reads中最小空间相对应的碱基。发生在minimizers之外的排序错误不会影响表示。minimizers中的那些会导致最小化空间中的替换或插入(图 4),可以使用 POA 识别并随后在minimizer space中矫正(图 1C)。
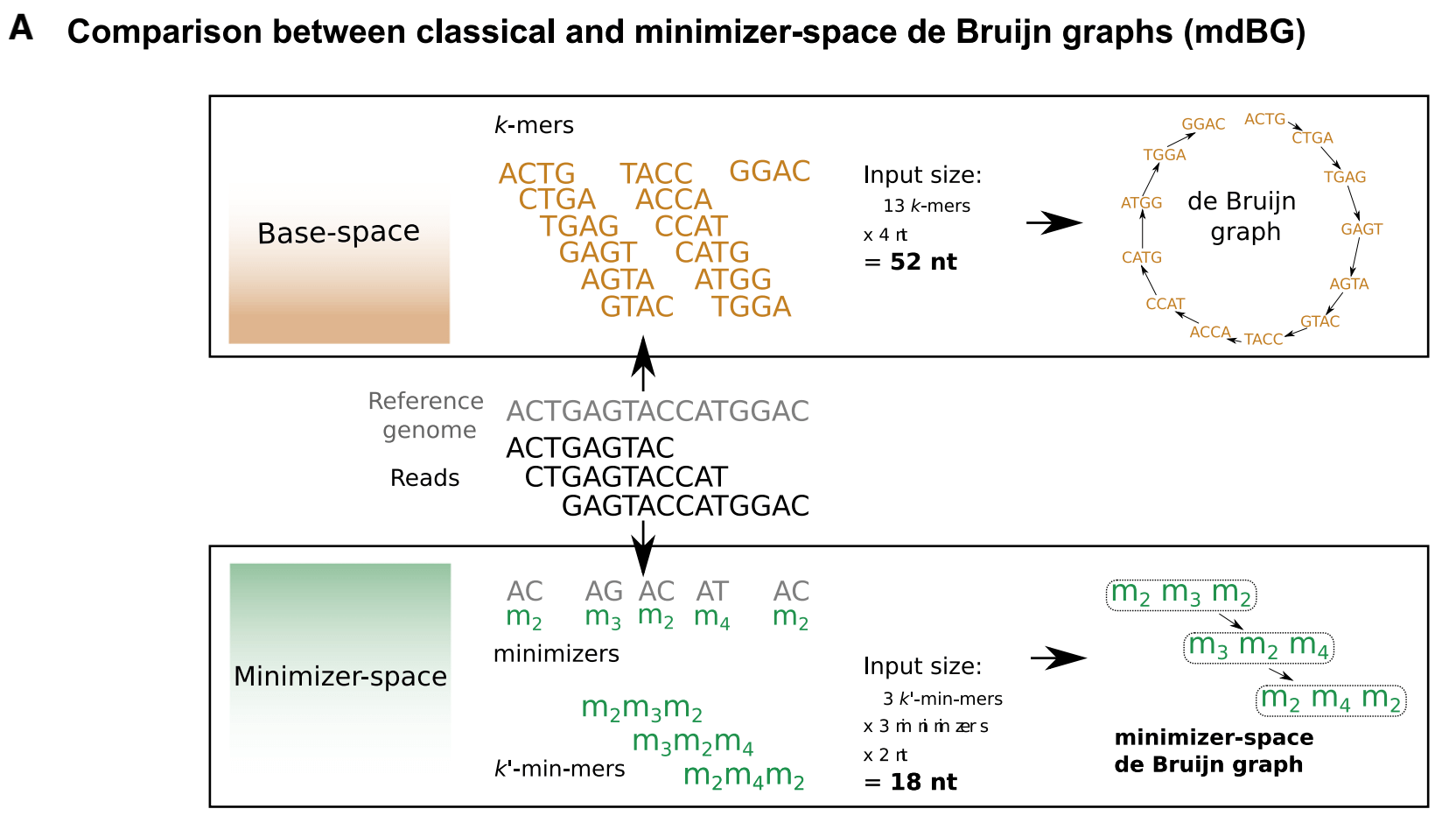
Fig 1A 一种用于基因组测序的高效组装方法(例如,PacBio HiFi数据)
minimizer-space de Bruijn graph(mdBG,底部)与通常用于基因组组装的原始de Bruijn graph(顶部)相比的图示
中间水平部分显示了一个参考基因组,以及测序reads。
从这幅图可以看出来,传统的Base-space方法是基于k-mers(k=4) 最终输入大小是52 nt 作者给出的 Minimizer-space方法,选择以核苷酸“A”开头的(AA, AG, AC, AT)作为最小值分别用m1, m2, m3, m4来表示。k’-min-mers(这里使用了k’=3,作为区分),从图中可以看出输入大小减少到18 nt。最小空间加快了de Bruijn图的构造和遍历,同时减少了内存消耗
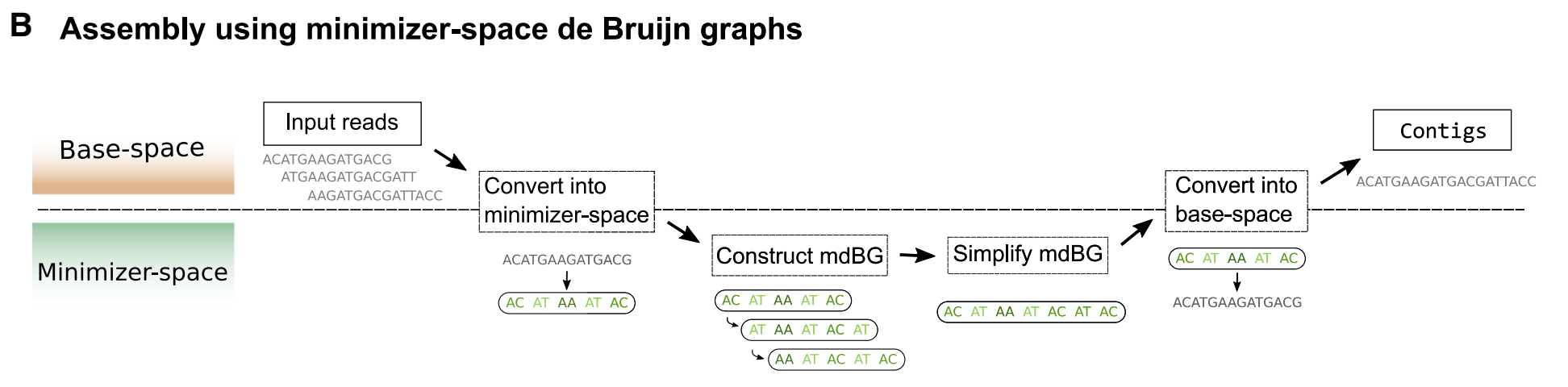
Fig 1B 使用mdBG的组装pipeline概述
两种方法虚线分开,上方按照顺序
虚线上方(下方) 的图形区域对应于在 基本空间(最小空间) 中进行的分析。
输入reads按顺序扫描,并识别所有属于预先选择的universe minimizers 的 δ-mers。 然后将每个reads表示为所选最小值的有序列表,并使用长度为 k 的滑动窗口从reads的最小空间表示中收集 k-min-mer。 然后从所有 k-min-mer 的集合构建最小空间 de Bruijn 图 (mdBG) 并进行简化,以减少歧义并消除错误。 然后通过连接由 mdBG 中的最小化器跨越的基空间序列,将 mdBG 转换回基空间,并报告一组contigs
通过使用mdBG执行组装,大大减少了向组装程序输入的数据量,保持了准确性,减少了运行时间,与当前的汇编程序相比,内存使用量减少了1到2个数量级。为de Bruijn图的阶数和最小化方案的密度设置适当的参数能够以与传统碱基空间组装类似的方式克服测序深度和read长度的随机变化。
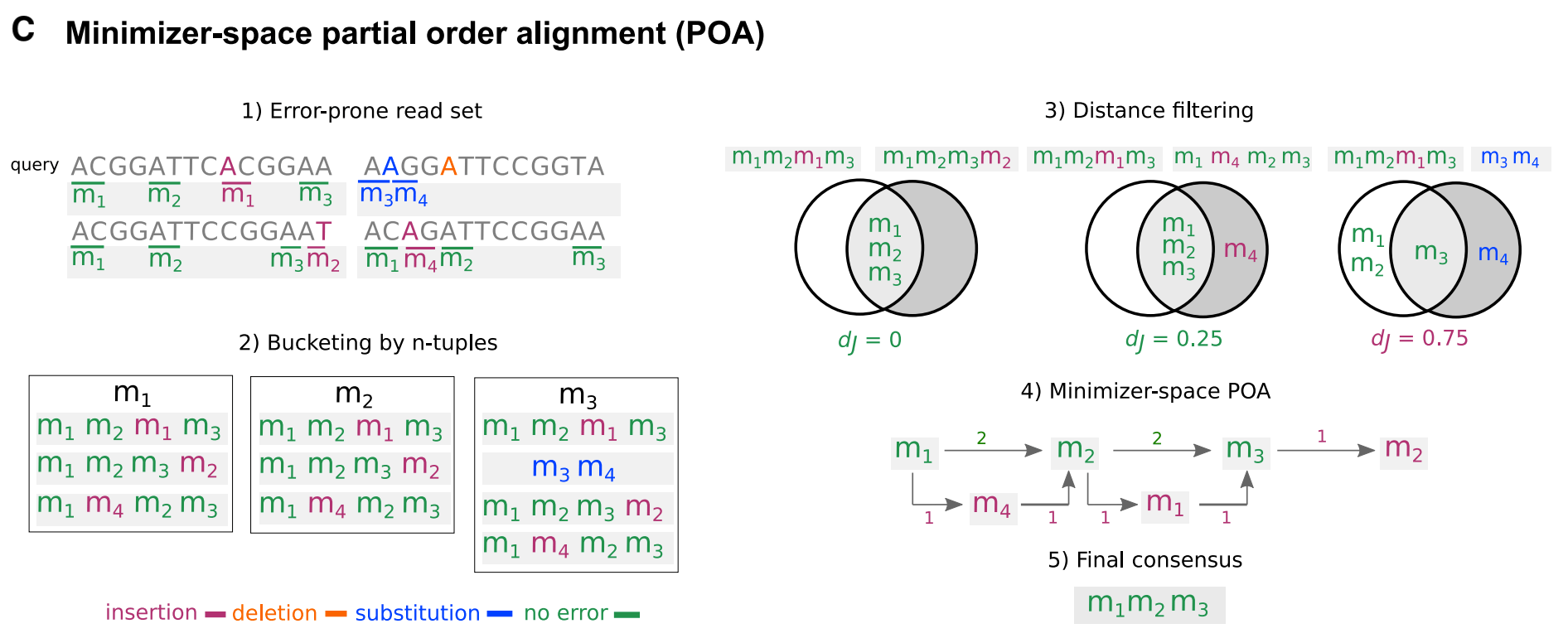
Fig 1C 具有 4 个reads的 toy 数据集的最小化空间偏序对齐 (POA) 过程概述
(1) 显示了容易出错的reads及其minimizers (δ = 2)的有序列表,带有排序错误和因错误而创建的最小化器以颜色表示(插入为红色,删除为橙色,替换为蓝色,绿色则表示没有错误)
(2) 在最小化空间纠错之前, minimizers的有序列表使用它们的 n-tuples (n = 1) 进行分组。
(3) 对于一个查询有序列表(图中read集合中的第一个read),得到所有与查询共享一个n-tuple的有序列表,通过启发式确定得到查询邻居的最终列表距离过滤器 (Jaccard 距离阈值 φ = 0.5)
(4) 最小化空间中的 POA 图是通过用查询初始化图并将通过过滤器的每个有序列表迭代地对齐到graph来构建(不太确定的边的权重显示为红色)
(5) 通过 graph 的共识路径,纠正查询中的错误。
为了处理更高的排序错误率,作者通过引入最小空间偏序对齐(POA) 的概念来纠正基本错误。
对于容易出错的数据,作者研究了两种情况:
果蝇和人类的PacBio HiFi reads数据(<1%错误率),由于率非常低,因此几乎不需要对错误进行调整;以及合成了错误率在1-10%的数据,对应于Nanopore数据的一个错误率范围。
作者还证明,尽管数据减少,但在合成的无错误数据和4%错误率数据上运行rust-mdbg软件可以实现近乎完美的基因组组装,后者(4%错误率)完全是因为在最小空间中应用了POA矫正的结果。
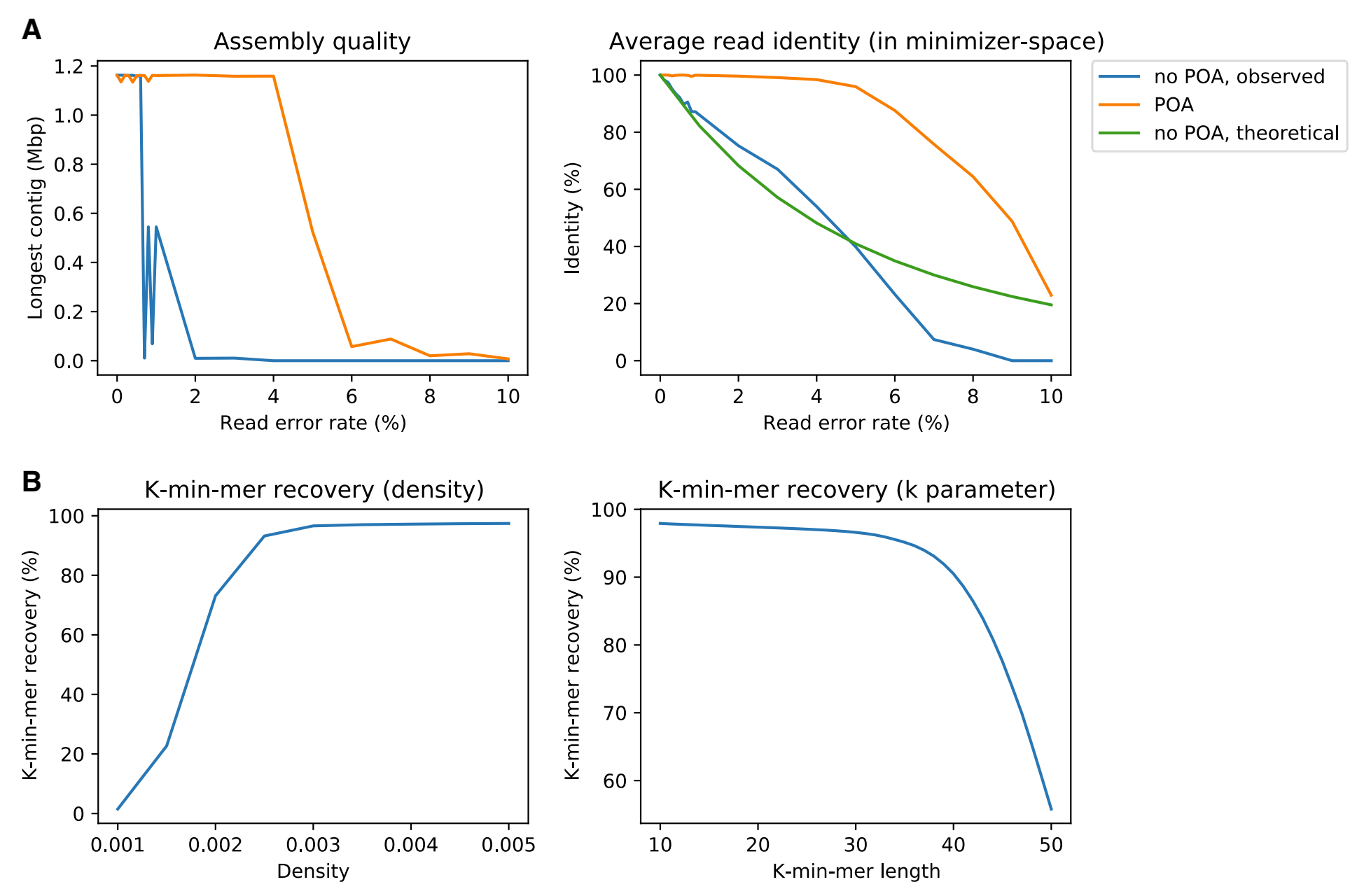
Fig 2 最小空间POA能够以更高的顺序错误率校正reads
作者引入最小空间偏序对齐(POA)来解决排序错误。为了确定最小空间POA的有效性以及具有较高read错误率的最小空间de Bruijn图组装的限制,作者在较小的数据集上进行了实验。
简言之,作者模拟了不同错误率下单个果蝇染色体的reads,并在有/无POA的情况下进行了mdBG组装。
图2A(左)显示,没有POA的原始实现只能将完整染色体重建为单个contig,错误率高达1%,之后染色体被组装成R2 contigs。使用POA,可以获得单个contig的精确重建,错误率高达4%。作者进一步验证,在3%的错误率下,除了reads中的基本错误外,重建的重叠群在结构上与参考完全对应。在错误率为4%的情况下,最小空间中的单个未修正索引会在组件中引入1 Kbp的人工插入。
图2A(右)表明,原始reads的最小空间随着错误率的增加而线性降低。使用POA,可以实现接近完美的校正,最高矫正误差率高达4%,误差率在>5%时急剧下降,但与未校正的reads相比,识别率仍然有所提高。
这突出了准确POA校正的重要性:从长远来看,mdBG似乎适用于没有POA的高保真数据(<1%错误率),作者的POA矫正几乎(但还不是完全)能够处理ONT数据的错误率(5%)。
使用POA时,实现运行时间约为45秒,内存为0.4 GB,而不使用POA时,运行时间小于1秒,内存小于30 MB。注意,作者没有使用优化的POA实现;因此,作者预计下一步的工作将显著降低运行时间,并可能提高校正质量。
为了进一步展示rust-mdbg的功能,作者使用它组装了两个PacBio HiFi宏基因组,实现了几分钟运行时间,以及使用较低的占用内存。比当前最先进的 hifiasm-meta 低两个数量级,组装完整性相当,但连续性较低。
作为最小空间分析的一个通用用例,作者构建了迄今为止661K细菌基因组中最大的泛基因组图,并在此图中对抗微生物耐药性(AMR)基因进行最小空间查询,识别出几乎所有与原始细菌基因组具有高度序列相似性的基因。
构建了迄今为止最大的 661K 细菌基因组泛基因组图,并在该图中执行对抗微生物耐药性 (AMR) 基因的最小化空间查询,鉴定几乎所有与原始细菌基因组具有高度序列相似性的基因。在大量样本中快速检测 AMR 基因将有助于实时 AMR 监测(Ellington 等,2017),而 mdBG 为索引 k-mer 搜索提供了一种节省空间的替代方案。值得注意的是,作者的方法相当于检查数据中输入碱基的可调比例(例如,只有 1%),并且应该推广到新兴的测序技术。
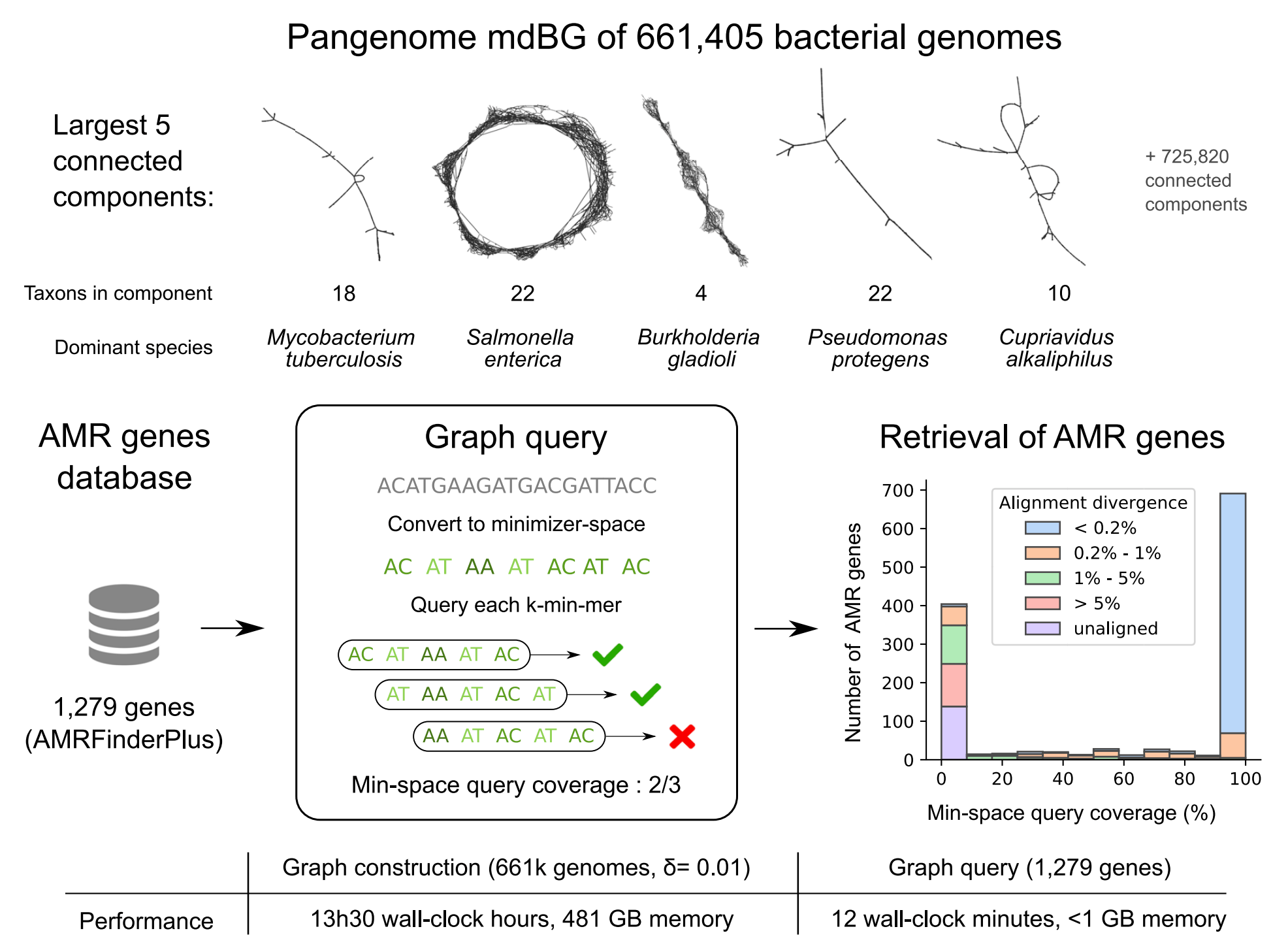
Fig 3 661405个细菌基因组的泛基因组mdBG和抗微生物耐药基因的检索
顶部面板:为整个661405细菌收集构建完整的 δ=0.001 泛基因组mdBG,并在此处显示前五个连接组件(使用Gephi软件)。每个节点是一个k-min-mer,边是k-min-mer之间 k - 1 最小值的精确重叠。
中间面板:将一组抗微生物抗性基因靶点转换为最小空间,然后在661405细菌泛基因组图(δ=0.01)中查询每个k-min-mer,产生基因检索的双峰分布:找到与泛基因组中的基因高度一致(99%+)的基因。柱状图由每个基因的最小序列趋异(sequence divergence)进行注释,该差异通过minimap2与其长度超过90%的泛基因组对齐得到。
底部面板:δ=0.01用于图形构造和查询的运行时和内存使用情况。注意,在预处理步骤中,该图只需构造一次。
实战结果比较
使用 rust-mdbg 对HiFi reads进行超快速、高效内存和高度连续的组装
作者在来自黑腹果蝇的PacBio HiFi reads上评估了作者的软件 rust-mdbg,覆盖率为 1003X,人类(HG002)的 HiFi reads为 503X,两者均取自HiCanu.
由于作者的方法不能解决二倍体生物体中的两种单倍型,作者与 HiCanu 和 hifiasm 的主要contigs进行了比较。在作者对黑腹果蝇的测试中,参考基因组由来自 RefSeq accession(GenBank:GCA_000001215.4) 的所有核染色体组成。使用 QUAST v5.0.2 进行组装评估,并使用 HiCanu 的文章中推荐的参数运行。 QUAST 将重叠群与参考基因组对齐,允许计算针对错误组装进行校正的连续性和完整性统计数据(表 3 中分别为 NGA 50和基因组分数指标)。程序集全部使用 8 个线程在 Xeon 2.60 GHz CPU 上运行。
对于 rust-mdbg 程序集,过滤掉短于 50 Kbp 的重叠群。没有报告基本空间转换步骤和图形简化的运行时间,因为它们不到运行 CPU 时间的 15% 并且在单个线程上运行,占用的内存不超过最终程序集大小,也比mdBG更少内存。
[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-KxKjiVBj-1635608738024)(https://z3.ax1x.com/2021/10/30/5ztD7n.png)]
表 1(最左侧) 显示了黑腹果蝇 HiFi 读取的组装统计数据。作者的软件 rust-mdbg 使用的时间比所有其他汇编程序少。在装配质量方面,所有工具都产生了高质量的结果。
表 1(最右侧) 显示了 Human HiFi (HG002) 读取的组装统计数据。
这突出表明 mdBG 允许在最小空间中对初始装配图进行非常有效的存储和简化操作。
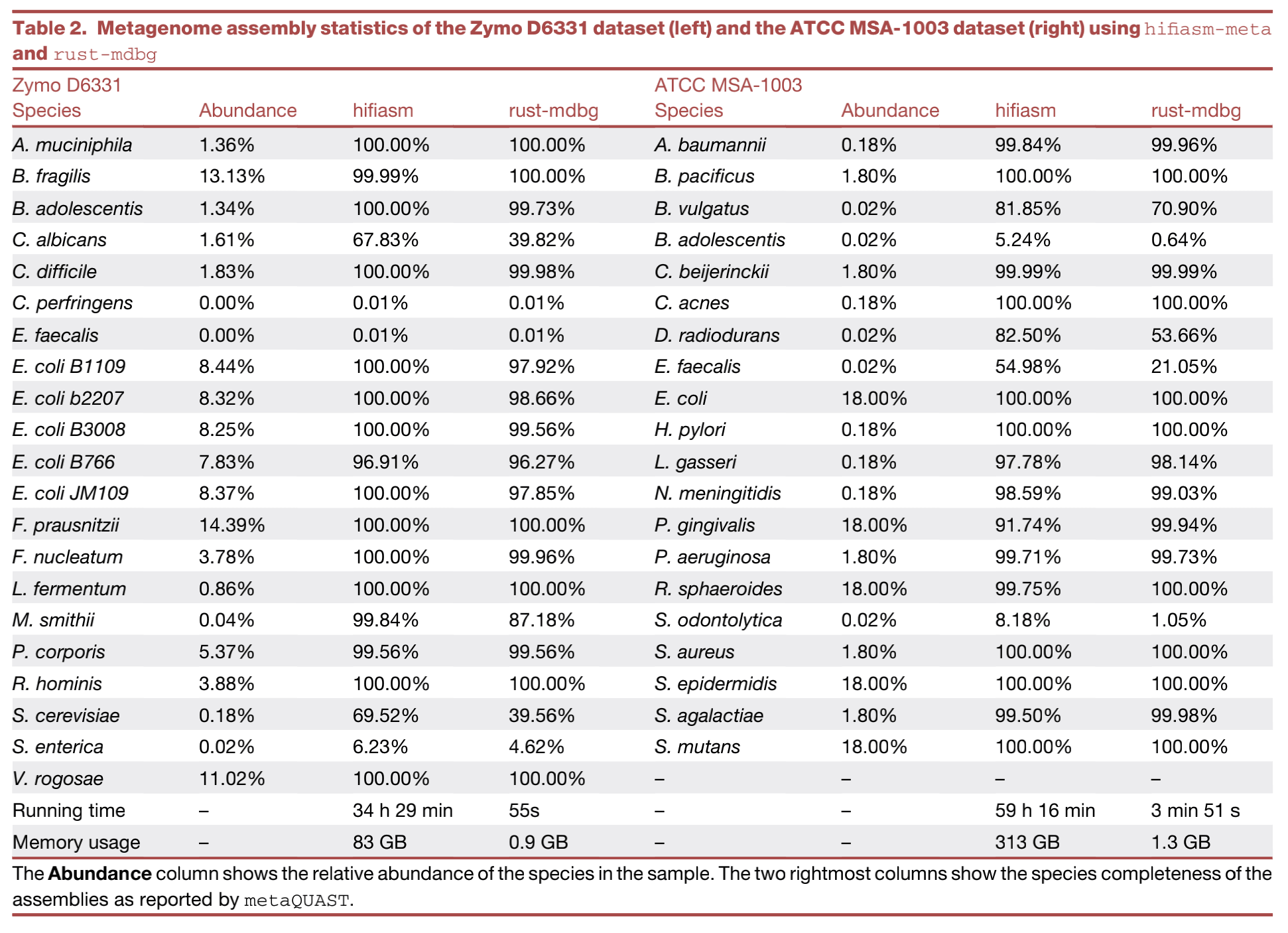

从表中可以看出运行时间以及运行内存都大大降低
但是要实事求是的讲,它的组装结果来说是没有其他结果的Complete(%)高,但是我觉得作者是从算法上的突破,让基因组的组装不在拘束于时间、内存、高配置服务器的限制,对于目前主流的组装软件来讲,当然不是一个很好的结果,但是也是一个突破。随着不断的完善进步,我相信可以既兼顾时间又兼顾组装的准确度。

Fig 4 base space到minimizer space的测序误差
作者考虑一个序列连同它的最小化器(图片的左边)。盒子里的每个面板描绘了不同突变对该序列的影响。
左上:G到C(紫色)导致最小空间表示没有变化,因为突变没有改变或产生任何最小空间。
左下:A到G导致m2消失。
右上:C到A使m3最小值出现。
右下:T到受影响的两个最小值:m4替换m1,插入m3。
总结
之所以分享这一篇文章,我想总结自从18年 Pacbio HiFi 测序技术出来之后,出现了很多hifi组装的软件,目前我认为结果较好的还是hifiasm软件,并且在多次的项目实践中证实了我的这一认为,当然还有很多优秀的组装软件比如:Canu、Falcon、Nextdenovo… 以及这次我介绍的这款软件,分享这款软件是因为它的算法进行了一个改变,使得组装时间大大缩短,并且也有一个不错的结果,作者目前更新的版本是v 1.0.1 ,可能有一些存在bug,等版本不断更新迭代肯定会更完善。 我相信,在不久的以后会有更多更好的算法以及软件会出现,降低了门槛,可以从基因组水平上对物种的生长、发育、进化、起源等重大问题进行研究,加深我们对物种的认识。
 微信
微信 支付宝
支付宝





