使用Ks方法进行全基因组复制事件研究
原理
全基因组复制(Whole Genome Duplication,WGD)是指在一次进化过程中整个基因组的复制。在植物的演化过程中,WGD是非常普遍的现象,它不仅影响着植物的基因组大小和结构,还对植物的形态、生态和适应性等方面产生了深远的影响。因此,对于植物WGD事件的研究具有重要的科学意义和实际应用价值。
Ks值是同源基因的替换率,通常被用来研究同源基因演化的速率。在全基因组复制事件发生后,同一个物种的基因组中同源基因的数量会翻倍,因此同源基因之间的Ks值峰值通常会向左移动。因此,Ks方法可以用来研究WGD事件的时间和方式等信息。
常用方法
在Ks分析中,同源基因序列被比对后计算其Ks值,通常使用剪枝法、最小二乘法或最大似然法等不同的计算方法。剪枝法是最常用的方法之一,它通过排除显然不合理的Ks值,从而提高计算的准确性和可靠性。
除了Ks分析,还有其他一些分子标记和生物信息学方法也可以用来研究WGD事件,如基于基因家族演化的分析方法、基于分子标记的时钟分析方法等。在具体研究中,不同的方法可以相互验证和印证,以提高分析结果的准确性和可靠性。
实例应用
Ks方法在植物基因组学和演化生物学研究中具有广泛的应用。例如,通过对不同物种中同源基因序列的Ks分析,可以推断它们之间的分化时间和进化历史。在研究WGD事件时,可以对同一个物种中的同源基因进行Ks分析,从而确定WGD事件的发生时间和方式。此外,还可以通过比较不同物种的Ks分布和峰值等特征来探究它们的进化关系和演化速率等问题。
总之,Ks方法是一种简单而有效的研究WGD事件的方法,它在植物基因组学和演化生物学研究中具有重要的应用价值和科学意义。在实际应用中,应结合其他方法进行相互验证和印证,
文件准备
分析流程和脚本均来自于此
首先将bin/ syn_analysis 文件夹以及 multispecies_ks2.py 和 synonymous_calc.py 与待分析物种的*.pep.fa 和 *.cds.fa 放在同一个目录下
注意:
*.pep.fa , *.cds.fa以及后面用cd-hit输出的 *.cd-hit 结果全都必须统一名称(包括序列名、文件名)
本文将使用示例数据黄芩(Scutellaria baicalensis )缩写为scubai 和 半枝莲(Scutellaria barbata )缩写为scubar 进行示范。
1 2 3 4 5 6 7 8 9 10 11 scubai.cds.fa scubai.pep.fa scubar.cds.fa scubar.pep.fa bin/ syn_analysis/ synonymous_calc.py multispecies_ks2.py
分析流程
使用CD-HIT去冗余
通过 CD-HIT (参数设置:-c 0.995 -n 5) 减少每个 .pep.fa 高度相似的序列
1 2 cd-hit -i scubai.pep.fa -o scubai.cd-hit -c 0.995 -n 5 -T 10 cd-hit -i scubar.pep.fa -o scubar.cd-hit -c 0.995 -n 5 -T 10
物种内Ks分析
运行
1 2 3 4 5 6 7 8 echo "scubai scubar" > name.txtpython2 /home/lixingze/scripts/wangning2020_portulacineae/sources/ks_plots.py 10 name.txt
物种间Ks分析
1 2 3 4 5 6 7 8 9 10 11 python multispecies_ks2.py num_cores target_taxon subject_taxa_list eg: python multispecies_ks2.py 10 A B,C,D,E python2 multispecies_ks2.py 10 scubai scubar
输出结果及可视化
输出结果为 ks.yn 以及 ks.ng 。任选一种即可,建议yn作为Ks分析的结果进行可视化
直方图
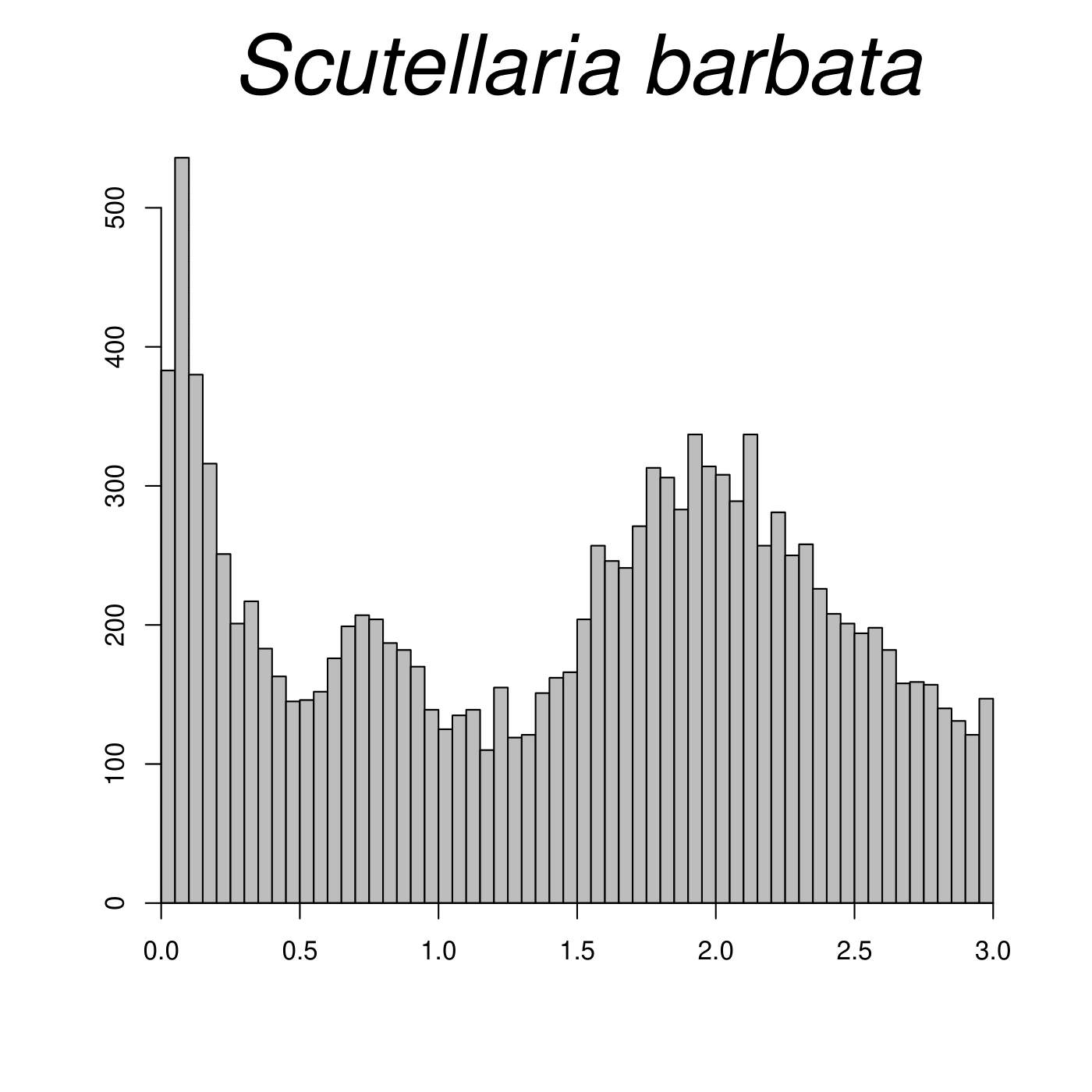
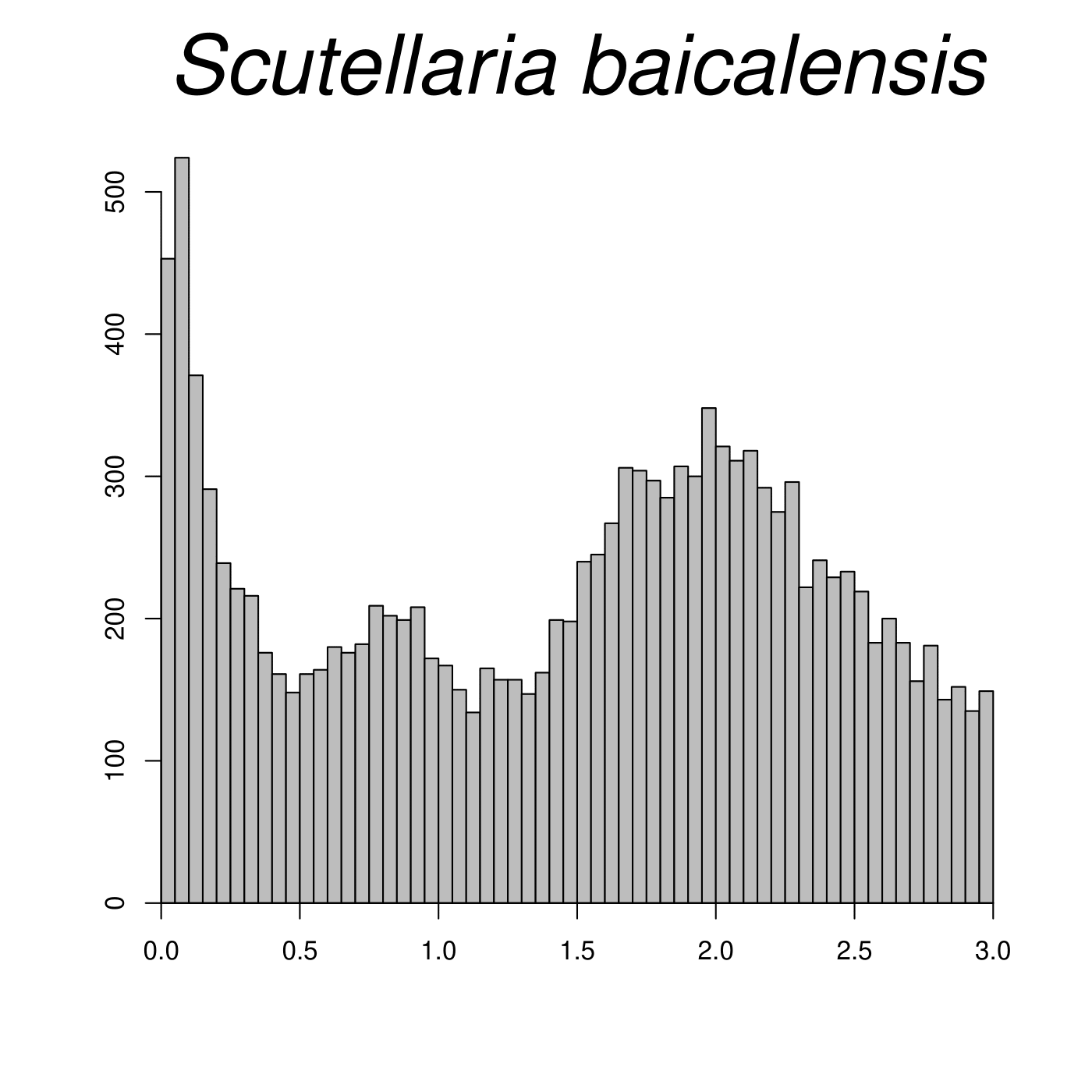
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 pdf=pdf(file='*.pdf' ,width=7 ,height=7 ) a<-read.table('*.ks.yn' ) a <- a[which(a > 0.02 ),] a <- data.frame(a) hist(a[,1 ],breaks=60 ,col='grey' ,xlab='' ,ylab='' ,main='your species name' , cex.lab=3 , cex.axis=3 , cex.main=3.2 , font.main=3 ,cex.sub=3 , axes=FALSE ) axis(1 ,pos=0 ) axis(2 ,pos=0 ) dev.off() pdf=pdf(file='scubai.ks.yn.pdf' ,width=7 ,height=7 ) a<-read.table('scubai.ks.yn' ) a <- a[which(a > 0.02 ),] a <- data.frame(a) hist(a[,1 ],breaks=60 ,col='grey' ,xlab='' ,ylab='' ,main='Scutellaria baicalensis' , cex.lab=3 , cex.axis=3 , cex.main=3.2 , font.main=3 ,cex.sub=3 , axes=FALSE ) axis(1 ,pos=0 ) axis(2 ,pos=0 ) dev.off() pdf=pdf(file='scubar.ks.yn.pdf' ,width=7 ,height=7 ) a<-read.table('scubar.ks.yn' ) a <- a[which(a > 0.02 ),] a <- data.frame(a) hist(a[,1 ],breaks=60 ,col='grey' ,xlab='' ,ylab='' ,main='Scutellaria barbata' , cex.lab=3 , cex.axis=3 , cex.main=3.2 , font.main=3 ,cex.sub=3 , axes=FALSE ) axis(1 ,pos=0 ) axis(2 ,pos=0 ) dev.off()
多物种比较图
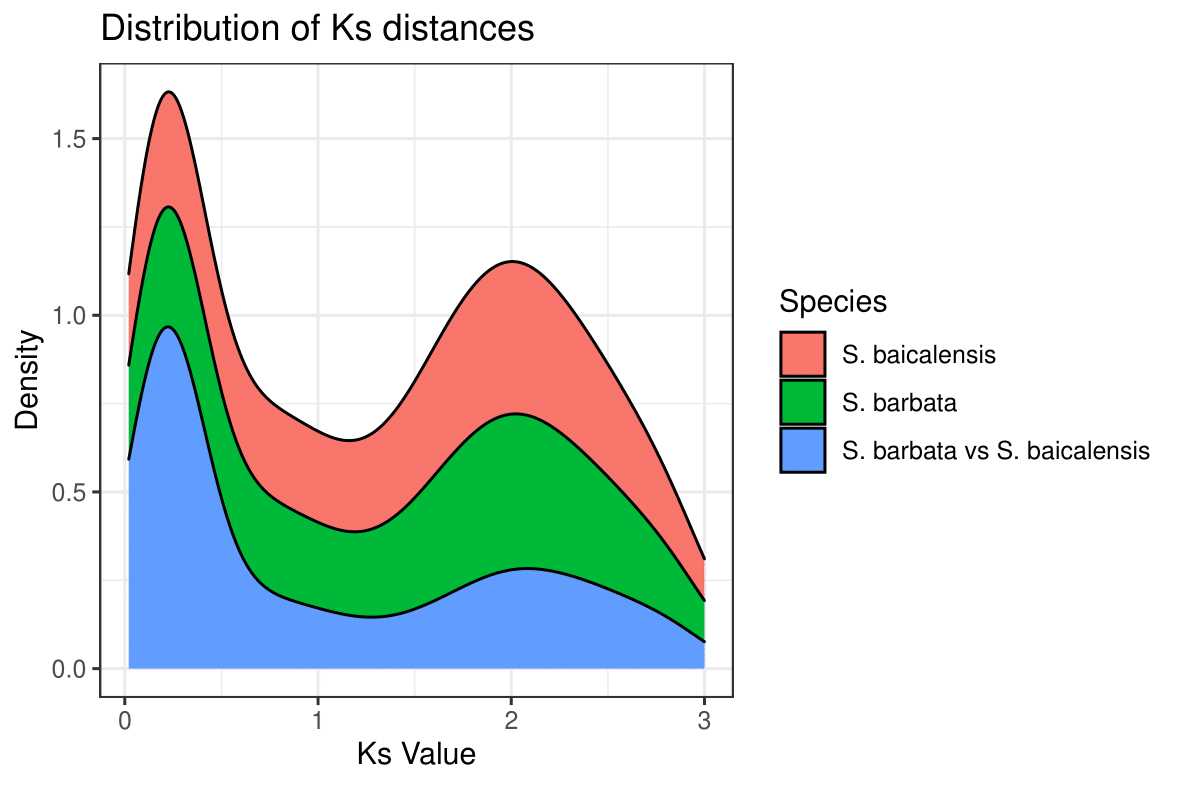
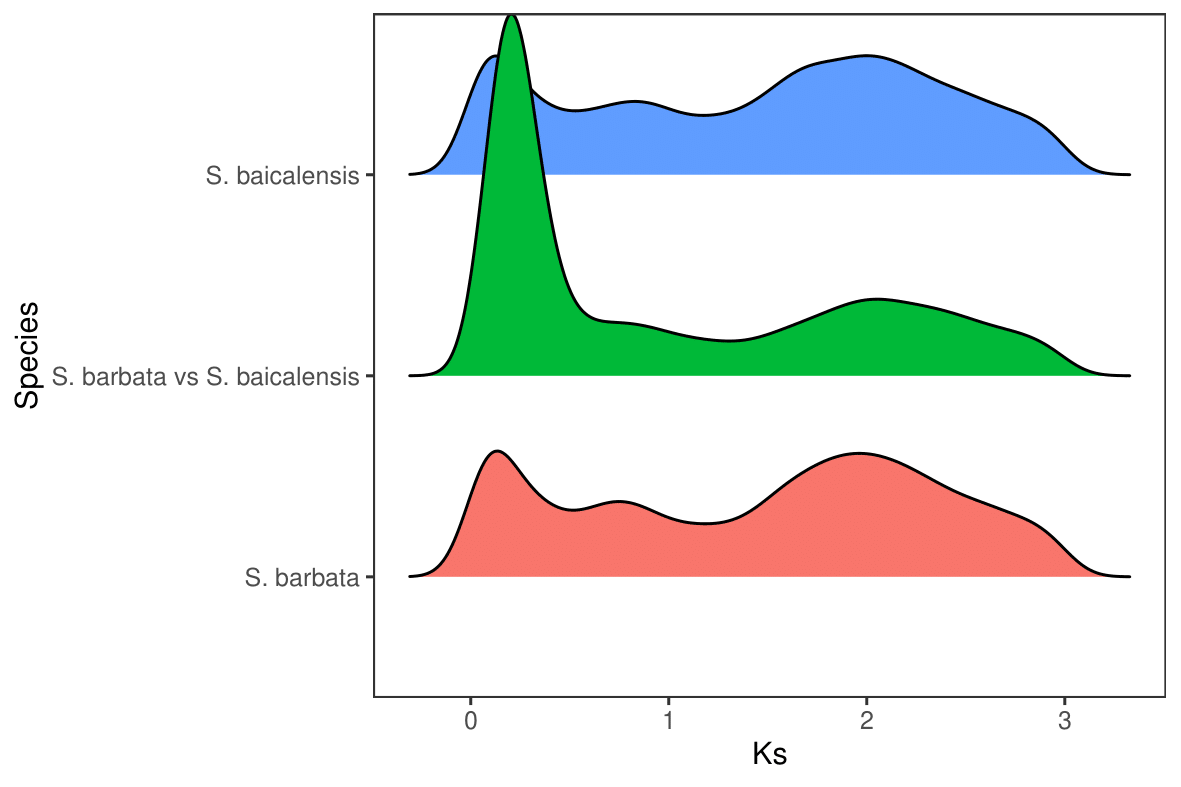
因为前面分析得到的Ks结果只有一列数字,如果要多个物种合并来看整体的Ks分布情况,需要在前一列加上物种名,以便后续区分。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 sed -i 's/^/S. barbata\t/g' scubar.ks.yn sed -i 's/^/S. baicalensis\t/g' scubai.ks.yn sed -i 's/^/S. barbata vs S. baicalensis\t/g' scubar_scubai.ks.yn cat *.ks.yn > all_species-ks.txt sed -i '1i\Species\tKs' all_species-ks.txt Species Ks S. barbata 0.2242 S. barbata 0.1898 S. barbata 0.3288 S. baicalensis 0.3732 S. baicalensis 0.059 S. baicalensis 0.0598 S. barbata vs S. baicalensis 0.4006 S. barbata vs S. baicalensis 0.4006 S. barbata vs S. baicalensis 0.2699
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 setwd("/Users/lixingze/Downloads/" ) data <- read.table("all_species-ks.txt" , header = T , check.names =F , sep = "\t" ) dtv <- subset(data,data$Ks>0.02 ) library(ggplot2) library(hrbrthemes) library(dplyr) library(tidyr) library(viridis) library(ggridges) p <- ggplot(data=dtv, aes(x=Ks, group=Species, fill=Species)) + geom_density(adjust=2 , alpha=1 , position="stack" ) + theme_bw()+ labs(title = "Distribution of Ks distances" , x = "Ks Value" , y= "Density" , size = 1.5 ) ggsave('out.pdf' ,height = 10 ,width = 15 ,units = 'cm' ,dpi = 600 ) p <- ggplot(data=dtv, aes(x = Ks, y = Species, fill = Species )) + geom_density_ridges() + theme( axis.title.x = element_text(face="bold" ), axis.title.y = element_text(face="bold" ), axis.text.y = element_text(face="bold.italic" , size = 10 ), axis.text.x = element_text(face="bold" , size = 10 ), legend.title = element_text(face="bold" , size = 10 ), legend.text = element_text(face="bold.italic" )) ggsave('out.pdf' ,height = 15 ,width = 25 ,units = 'cm' ,dpi = 600 )
 微信
微信 支付宝
支付宝





