OrthoFinder 进行直系同源基因分析教程
介绍
OrthoFinder 是一个快速、准确和全面的比较基因组学平台。 它找到正交群(orthogroups)和直系同源(orthologs),推断所有正交群的有根基因树,并识别这些基因树中的所有基因复制事件。它还为被分析的物种推断出一个有根的物种树,并将基因复制事件从基因树映射到物种树的分支。
OrthoFinder 还为比较基因组分析提供全面的统计数据。 OrthoFinder 使用简单,运行它所需的只是一组 FASTA 格式的蛋白质序列文件(每个物种一个)。
总的来说,它将要分析的物种的蛋白质组作为输入,并从这些蛋白质组中:
- 推断目标物种的正交群
- 推断出一组完整的有根基因树
- 推断有根物种树
- 使用基因树推断基因之间的所有直系同源关系
- 推断基因复制事件并将它们交叉引用到基因和物种树上的相应节点
- 为目标物种提供比较基因组学统计数据
除了大规模分析外,它还可以用于在实验研究之前仔细检查各个直系同源关系。
安装
使用Conda安装
1 | conda install orthofinder |
本地安装
可以使用 Bioconda 安装 OrthoFinder 或直接从 GitHub 下载。
1 | # 从github下载最新版本 |
命令行选项
开始分析的选项
1 | -f <dir>:从 FASTA 文件目录开始分析 |
停止分析的选项
1 | -op:在为全对全序列搜索准备输入文件后停止(例如 BLAST/DIAMOND) |
控制工作流程的选项
1 | -M <opt>:使用 MSA 或 DendroBLAST 基因树推断,opt=msa,dendroblast [默认=dendroblast] |
控制所用程序的选项
1 | -S <opt>:序列搜索程序 opt=blast,diamond,mmseqs,... 用户可扩展 [默认 = 菱形] |
更多选项
1 | -d:输入是 DNA 序列 -t <int>:用于序列搜索、MSA 和树推理的线程数 [默认为机器上的内核数] |
分析前的准备
物种的选择
三个标准分析是:
- 跨物种进化枝进行比较分析
- 识别一对或少数物种之间的直系同源物
- 研究进化史上特定点的变化
在第一种情况下,只需尽可能获取进化枝中所有物种的pep序列。通常,不需要为感兴趣的进化枝包含一个外群。因为这会推迟进化历史中定义正交群(Orthogroups, Orthologs & Paralogs)的点,因此最好不要,如果添加外群的话正交群将具有较低的分辨率。
在第二种情况下,最好确保有足够的物种以获得最佳结果。同样的规则适用于推断一个好的系统发育树:应该用中间物种分解长分支。需要最少 4 个物种,最佳选择6-10 个。
如果对物种树特定分支上发生的事感兴趣,那么同样应该确保良好的物种选择。理想情况下,分支下方至少有两个物种,上方最近的分支至少有两个物种,以及在外群至少有两个物种。
转录组和低质量基因组
一般来说,最好使用可用的最佳注释基因组,但 OrthoFinder 对缺失基因非常稳健,因此这不是一个大问题。 转录组可能出现的一个问题是,当从每个物种大约 100,000 个转录本开始时。 这在计算上可能很费时,并且可能会导致生成大量文件,因此在这种情况下要多注意。
使用哪个proteome版本
OrthoFinder 使用氨基酸序列作为蛋白质编码基因。 理想的情况是对每个基因使用单一的 primary/longest transcript variant
这也将大大减少运行时间。
- Ensembl:使用
.pep.all.fa文件而不是.pep.abinitio.fa,据我所知,这些是更好的支持基因模型(如果有问题请纠正)。通常,不是每个基因只有一个代表性的转录本,但有一个随 OrthoFinder 一起提供的脚本提取每个基因的最长转录本,推荐使用它。
Ensmbl 上还有用于下载基因组的子站点:bacteria、protists、fungi、metazoa
- Phytozome使用
.protein_primaryTranscriptOnly.fa文件
物种名称
OrthoFinder 将使用每个蛋白质组文件名作为该物种的名称。使用这些的地方是在基因树中,其中每个基因名称都以其物种名称为前缀。这非常有助于解释需要查看的任何基因树,因为如果节点是重复或物种形成事件,它会显而易见。它还可以查看基因树中的基因是否以期望它们获得物种树知识的方式相关。
因此,使用简洁的命名风格。例如,对来自 Phytozome 的植物基因组运行 OrthoFinder 时,给文件命名为A_thaliana.fa和O_sativa.fa。
基因名称
OrthoFinder 将通过测试每个登录行上的第一个(空格分隔的)单词是否唯一,尝试找到一组简短的、唯一的基因名称来引用每个序列。
如果是,那么基因将通过这些名称来识别。否则,将使用完整的登录行来指代每个基因。由于the quadratic nature of orthology(每个物种对一个直系同源结果文件),每个基因名称将被写出 O(n) 次,因此对于大型分析,使登录行整洁将大量节省磁盘空间以及 OrthoFinder 写出所有直系同源结果文件所花费的时间!
如果将上述脚本用于 Ensembl proteomes,那么文件将被正确解释,因此它们具有合适的格式,每个序列都有一个基因标识符。
运行
示例数据运行
1 | OrthoFinder/orthofinder -f OrthoFinder/ExampleData |
实际运行
自己的数据集的话需要将OrthoFinder/ExampleData替换为你的蛋白序列文件所在的文件夹位置。
OrthoFinder 将查找具有以下任何文件扩展名的输入 fasta 文件:
1 | .fa |
结果文件解析
默认情况下,OrthoFinder 在输入目录中创建一个名为“OrthoFinder”的结果目录,并将结果放在此处。结果目录如下所示:
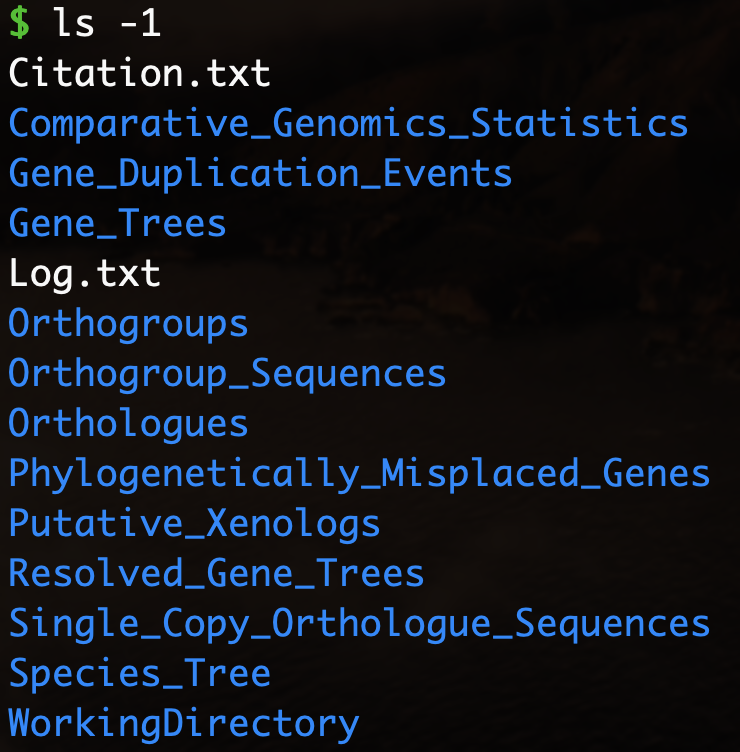
测试数据的结果文件下载
1 | wget https://bioinformatics.plants.ox.ac.uk/davidemms/public_data/Results_model_species.tar.gz --no-check-certificate |
Phylogenetic Hierarchical Orthogroups
从 2.4.0 版本开始,OrthoFinder 通过分析有根基因树来推断每个层级(即物种树中的每个节点)的 HOG、正交群。
这是一种比以前由 OrthoFinder 使用的基于基因相似性/图形的方法(已弃用的 Orthogroups/Orthogroups.tsv 文件)更准确的正交群推断方法。
根据 Orthobench benchmarks,这些新的正交群比 OrthoFinder2 正交群 (Orthogroups/Orthogroups.tsv) 准确 12%。通过包括外群物种,可以进一步提高准确度(在 Orthobench 上准确度提高 20%),这有助于解释有根基因树。
-
N0.tsv
是制表符分隔的文本文件。每行包含属于单个正交群的基因。来自每个正交群的基因被组织成列,每个物种一个。额外的列给出了 HOG(分层正交群)ID 和基因树中确定 HOG 的节点(注意,这可以在包含基因的进化枝的根之上)。该文件有效地替换了使用 MCL 进行马尔可夫聚类的Orthogroups/Orthogroups.tsv 中的正交群。 -
N1.txt, N2.tsv, …
Orthogroups 从与物种树 N1、N2 等中物种进化枝对应的基因树推断出来。现在可以在分析中包含外群物种,然后使用 HOG 文件获取为物种树中所选进化枝定义的正交群。
(分层正群分裂:在分析基因树时,嵌套的分层组(除 N0 以外的任何 HOG,所有物种的最后一个共同祖先级别的 HOG)有时可能会丢失最早发散物种的基因,然后复制在第一个现存基因之前。即使有证据表明它们属于同一个 HOG,但两个最初的分歧进化枝将是旁系同源的。对于大多数分析,通常最好将这些进化枝分成单独的组。这可以使用选项 ’ 请求-y '.)
Species Tree 文件夹
进化树查看软件(有很多简单列举几个我用到的)
-
Denroscope是一个树查看器,可以下载并在本地运行,如果要查看不止几棵树,它是最佳选择。
-
Figtree 一种用于进化生物学的进化树作图软件,主要用于生物进化系统树,并且支持多种形式的进化树,支持有颜色、名称变化等功能
-
从 Web 浏览器查看,例如 ETE Toolkit tree viewer。
使用其中之一,打开文件Species_Tree/SpeciesTree_rooted.txt。由于此文件有bootstrap值,Denroscope 需要选择 Interpret as edge labels 选项才能正确查看它们。物种树看起来像这样:
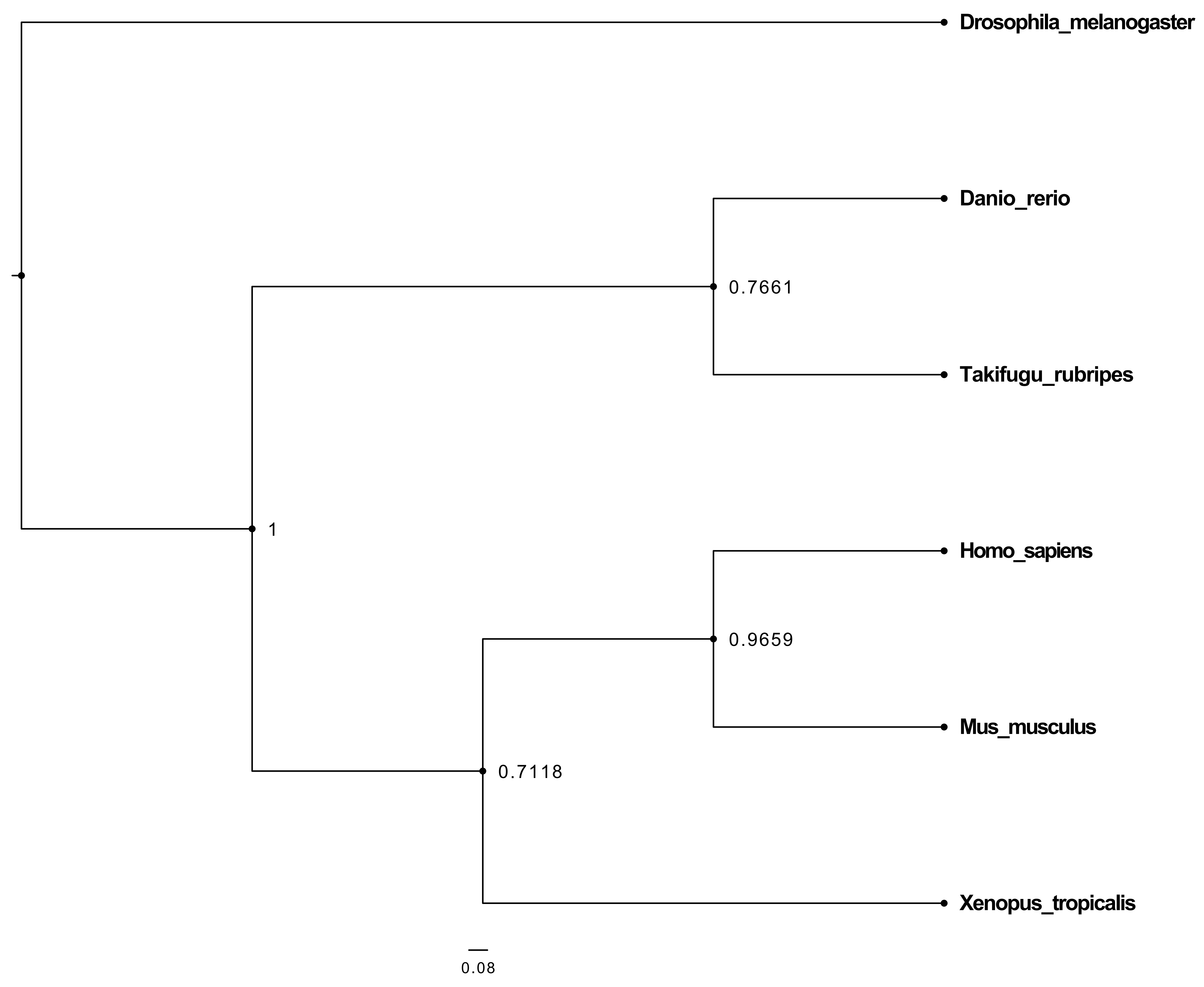
这棵树是由 OrthoFinder 使用STAG算法推断出来的,并使用STRIDE算法进行了生根。
如上所述,可以在此处看到果蝇位于比其他物种更长的分支上。如果知道物种树应该是什么样子,应该检查树是否符合预期。这里推断树是正确的。
如果物种树不正确,也不会影响 orthogroup 推断,但它可能会影响某些具有基因重复事件的基因树中的 orthologue 推断。在这种情况下,可以使用更正后的物种树(-ft和-s选项)再次运行 OrthoFinder 分析。通常运行很快,因为所有计算量大的计算(正交群和基因树的推断等)都已经完成。
在这棵树中,支持值并非都是 100%。
使用默认选项,物种树推断是使用 STAG 执行的,它使用从支持每个二分的单基因座基因树派生的物种树的比例作为其支持的度量。 这是比多序列比对的标准引导程序支持更严格的措施。
如果改为使用-M msa选项,则将使用串联的多序列比对代替物种树推断,并且所有二分法的支持度值为 100%。 在这种情况下,支持值对应于从完整的多基因比对中获取的引导复制,这是完全不同的事情。 这是最常用的支持度量,对于相同的数据总是会报告更高的支持值。
-
SpeciesTree_rooted.txt
从所有正交群推断的 STAG 物种树,包含内部节点处的 STAG 支持值并使用 STRIDE 植根。 -
SpeciesTree_rooted_node_labels.txt
与上面相同的树,但节点被赋予标签(而不是支持值),以允许其他结果文件交叉引用物种树中的分支/节点(例如基因复制事件的位置)。
Orthologues
运行 OrthoFinder 的最常见原因之一是找到感兴趣的直系同源基因
实际的操作流程如下:
- 找一个感兴趣的基因比如:看看果蝇基因
FBgn0005648[它参与核裂解/聚腺苷酸化反应的裂解和聚腺苷酸化步骤(见FlyBase)] 的 orthologues
在Orthologues目录中,每个物种都有一个子目录,该子目录又包含每个成对物种比较的文件,列出该物种对之间的直向同源物。
打开Orthologues/Orthologues_Drosophila_melanogaster/Drosophila_melanogaster__v__Homo_sapiens.tsv
该文件包含三列,Orthogroup、Drosophila_melanogaster和Homo_sapiens。在表中找到FBgn0005648,会看到该基因在正交群OG0001189中,并且它在人类中具有三个orthologues:ENSG00000205022、ENSG00000100836、ENSG00000258643。
Gene Trees
为具有 4 个或更多序列(4 个序列是大多数树推理程序进行树推理所需的最小数量)的每个正交群推断出的有根系统发育树。
在上面发现FBgn0005648在人类中具有三个直系同源基因,接下来我们将查看基因树,看看是否符合这一点,看看这三个直系同源物是如何产生的。
打开Gene_Trees/OG0001189_tree.txt
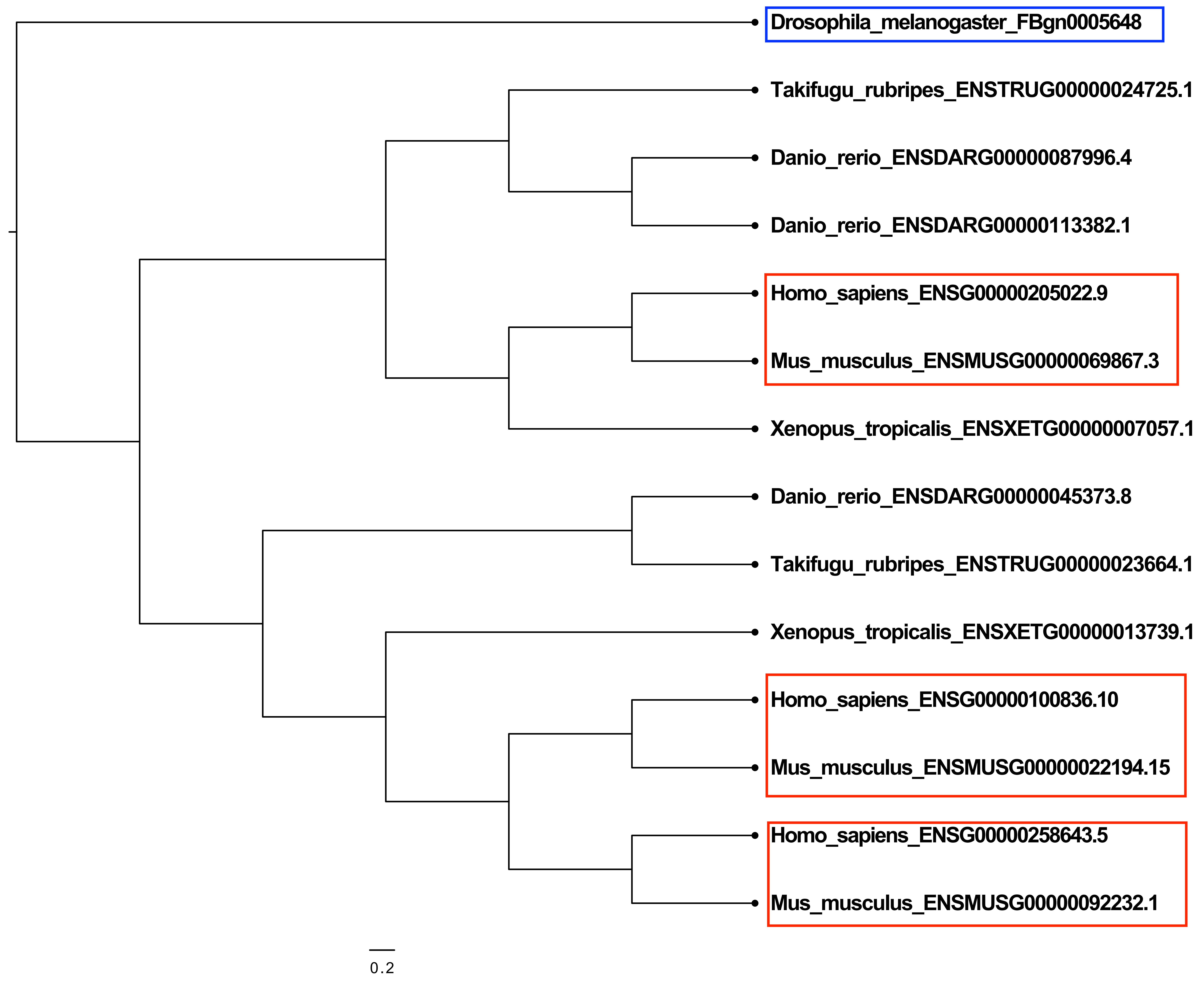
从图片可以看出,OrthoFinder 已自动植根 ———— 这棵树植根于Drosophila基因 FBgn0005648。这使得快速检查基因树非常方便,对于更复杂、更难解释的基因树尤其有用。
查看基因树,我们可以看到发生了两个基因复制事件,一个由脊椎动物共享,另一个由人和小鼠共享。这导致了一对三的直系同源关系,即所有三个人类基因都与一个果蝇基因密切相关。通常情况下,直系同源关系不是一对一的,了解这一点很重要(如果不想花几个月的时间对“直系同源”进行实验)
我们可以在 FlyBase 上查看这个基因的页面:http://flybase.org/reports/FBgn0005648.html
转到Orthologs部分,然后查看Human Orthologs,会发现识别所有这三种直向同源物的方法是基于树的方法 Compara、eggNOG、OrthoFinder 和 TreeFam。OrthoFinder 是唯一可以在自己的数据上运行的工具。基于评分的方法,例如 Hieranoid、Inparanoid、OMA 和 OrthoMCL,仅识别出这些直向同源物中的一个或一个。基因树对于识别和解决这些复杂的关系尤为重要。
默认基因树没有支持值。毕竟,OrthoFinder 已经将大约 121,000 个基因分配到正交群中,并在大约 15 分钟内为这些基因推断了近 18,000 个基因树!我们将在后面的教程中讨论如何获取支持值。
Gene Duplication Events
拥有基因树意味着 OrthoFinder 可以识别发生的所有基因复制事件。
OrthoFinder 在文件Species_Tree/ SpeciesTree_rooted_node_labels.txt 中标记物种树的节点
查看节点N1,脊椎动物的共同祖先(即 D. rerio、T. rubripes、X.tropicalis、H. sapiens和M. musculus)。有两个文件提供了有关基因复制事件的详细信息。我们先打开Gene_Duplication_Events/SpeciesTree_Gene_Duplications_0.5_Support.txt
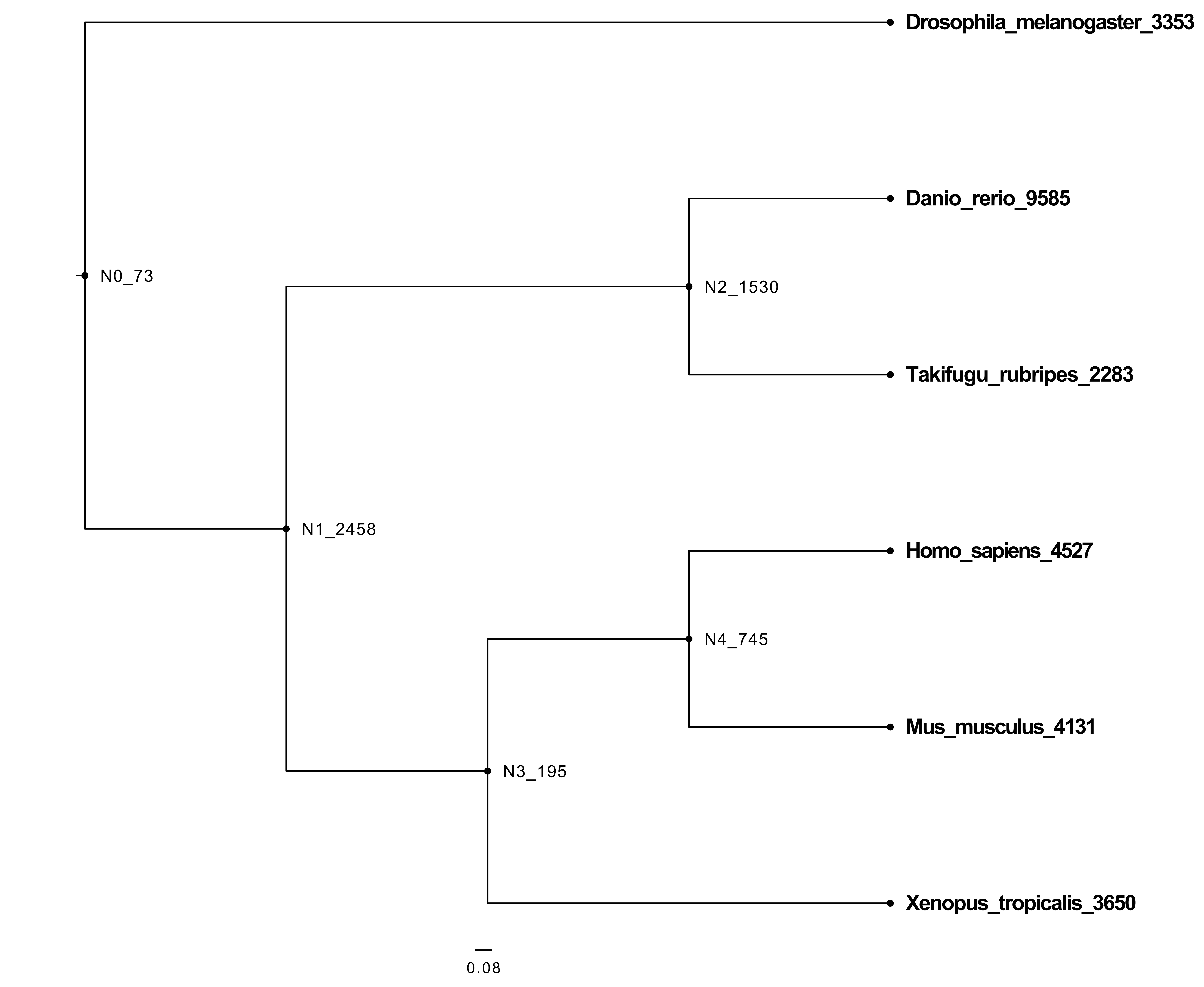
以上给出了基因复制事件的Summary。其中每个节点显示节点名称,后跟一个下划线,然后是映射到物种树中每个节点充分支持的基因复制事件的数量。
如果至少 50% 的后代物种保留了复制基因的两个拷贝,则基因复制事件被认为是“得到充分支持的”。
对于四足动物的共同祖先 N1,有 2458 个得到充分支持的基因复制事件。
我们可以在文件Gene_Duplication_Events/Duplications.tsv 中看到这些基因复制事件的列表。以下是文件中的几行,按它们发生的物种树节点排序:
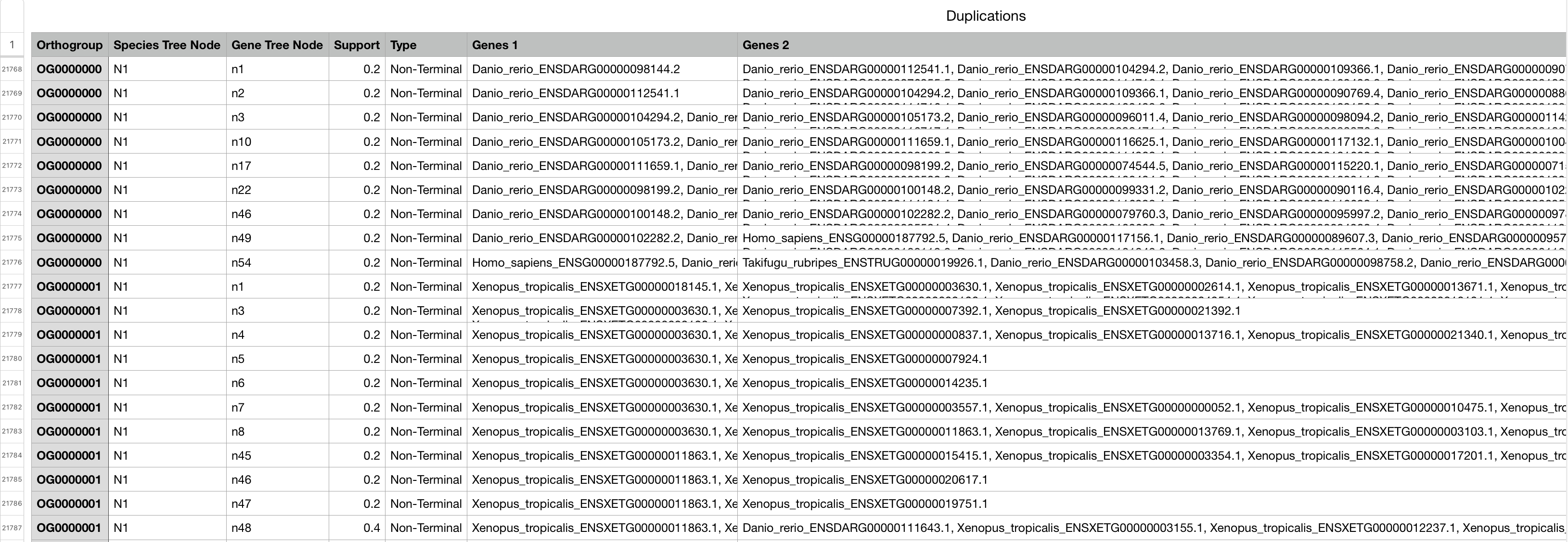
每个基因复制事件都与物种树节点、它发生的正交群/基因树和该基因树中的节点交叉引用。
-
Duplications.tsv
是一个制表符分隔的文本文件,它列出了通过检查每个正群基因树的每个节点识别出的所有基因复制事件。列是“Orthogroup”,“Species Tree node”(发生复制的物种树的分支,参见Species_Tree/SpeciesTree_rooted_node_labels.txt),“Gene tree node”(与基因复制事件对应的节点,参见相应的orthogroup Resolved_Gene_Trees/) 中的树;“支持”(存在复制基因的两个副本的预期物种的比例);“类型”(“终端”:物种树终端分支上的重复,“非终端”:物种树内部分支上的重复,因此被多个物种共享,“非终端:STRIDE检查基因树的拓扑结构在复制后应该是什么);“基因 1”(基因列表来自复制基因的一个副本),“基因 2”(基因列表来自复制基因的另一个副本。 -
SpeciesTree_Gene_Duplications_0.5_Support.txt
提供了物种树分支上的上述重复的总和。它是一个 newick 格式的文本文件。每个节点或物种名称后面的数字是在导致节点/物种的分支上发生的具有至少 50% 支持度的基因复制事件的数量。分支长度是标准分支长度,如 Species_Tree/SpeciesTree_rooted.txt 中给出的。
Resolved Gene Trees
Orthofinder还列出了从基因复制事件产生的两个拷贝中的每一个的后代的基因。
我们可以查看 FBgn0005648 直系同源物。
通过 Resolved_Gene_Trees/OG0001189_tree.txt 中的基因树。 该目录包含带有标记节点的基因树。
这些文件显示了 OrthoFinder 在推断直向同源物和基因复制事件时如何解释基因树。 它们可能与直接来自tree inference 步骤(在 Gene_Trees/ 中可用)的原始基因树略有不同。 为了获得已解析的基因树,OrthoFinder 进行了重复-丢失-合并分析,以确定对基因树的更简洁的解释。
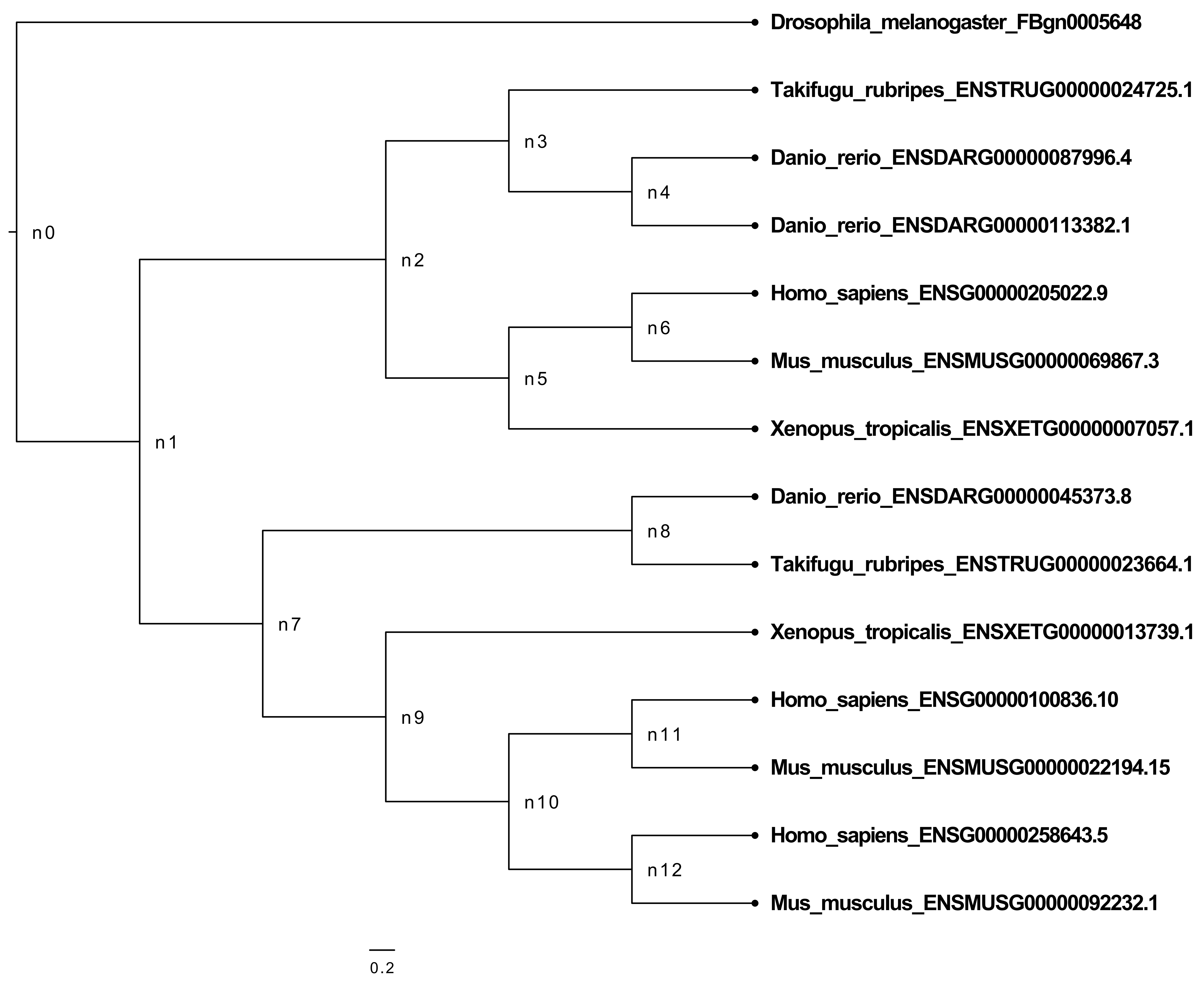
从表中可以看出,在节点 n1 处发生了基因复制事件,所有后代物种中的两个副本均 100% 保留。 查看树,第二个基因复制事件发生在节点 n10 上,如果我们回到表格中,我们可以看到这个列表以及 Danio rerio 中的终端基因复制事件:

如果对基因复制事件感兴趣,那么此表包含大量数据。 在这六个物种中,OrthoFinder 确定了 34,065 个基因重复事件,所有这些事件都与它们发生的物种树和基因树的节点交叉引用! 这些事件也按正交群和物种树节点汇总在文件 Duplications_per_Orthogroup.tsv 和 Duplications_per_Species_Tree_Node.tsv 中,它们都在目录 Comparative_Genomics_Statistics/ 中。
为具有 4 个或更多序列的每个正交群推断出有根的系统发育树,并使用 OrthoFinder hybrid species-overlap/duplication-loss coalescent模型进行解析。
Comparative Genomics Statistics
-
Duplications_per_Orthogroup.tsv
一个制表符分隔的文本文件,它给出了每个正交群中标识的重复数。此数据的主文件是 Gene_Duplication_Events/Duplications.tsv。 -
Duplications_per_Species_Tree_Node.tsv
它给出了识别为沿着物种树的每个分支发生的重复数。此数据的主文件是 Gene_Duplication_Events/Duplications.tsv。 -
Orthogroups_SpeciesOverlaps.tsv
包含作为方阵的每个物种对之间共享的正交群的数量。 -
OrthologuesStats_*.tsv
包含矩阵给出每对物种之间一对一、一对多和多对多关系中的直向同源物数量。 -
OrthologuesStats_one-to-one.tsv
是每个物种对之间一对一直向同源物的数量。 -
OrthologuesStats_many-to-many.tsv
包含每个物种对的多对多关系中的直向同源物的数量(由于物种形成后两个谱系中的基因重复事件)。条目 (i,j) 是物种 i 中与物种 j 中的基因存在多对多直系关系的基因数。 -
OrthologuesStats_one-to-many.tsv
条目 (i,j) 给出物种 i 中与物种 j 的基因处于一对多直系关系的基因数量。这里有一个示例结果文件的演练 (https://github.com/davidemms/OrthoFinder/issues/259)。 -
OrthologuesStats_many-to-one.tsv
条目 (i,j) 给出物种 i 中与物种 j 中的基因处于多对一直系关系的基因数量。 -
OrthologuesStats_Total.tsv
包含任何多样性的每个物种的直向同源物对的总数。条目 (i,j) 是物种 i 中在物种 j 中具有直向同源物的基因总数。 -
Statistics_Overall.tsv
是一个制表符分隔的文本文件,其中包含有关正交群大小和分配给正交群的基因比例的一般统计信息。
总的来说,至少 80% 的基因被分配到正交群。少于此值意味着可能会遗漏某些剩余基因实际存在的直系同源关系,物种采样不佳是造成这种情况的最可能原因。 让我们也检查每个物种的百分比。 -
Statistics_PerSpecies.tsv
是一个制表符分隔的文本文件,它包含与 Statistics_Overall.csv 文件相同的信息,但针对每个单独的物种。
Orthogroups
每个正交群的 FASTA 文件给出了正交群中每个基因的氨基酸序列。
通常我们对分组物种比较感兴趣,即跨物种进化枝(而不是一对物种之间的比较)。orthogroup 对多个物种的泛化是orthogroup。就像直系同源物是一对物种的最后一个共同祖先中的单个基因的后裔一样,正交群是一组物种中的单个基因的后裔。OrthoFinder 中的每个基因树,例如上面的基因树,都对应一个正交群。如果我们希望它包含所有成对直向同源物,那么正交群基因树就是我们需要查看的树。并且即使正交群中的某些基因可以是彼此的旁系同源物,但如果我们试图去除任何基因,那么我们也将移除直系同源物。
因此,如果我们想对一组物种中的“等效”基因进行比较,我们需要对同种群中的基因进行比较。正交群位于文件Orthogroups/Orthogroups.tsv 中。该表每行有一个正交群,每列有一个物种,并从最大的正交群到最小的正交群排序。还有一个传统 OrthoMCL 格式的文件:Orthogroups/Orthogroups.txt。
Single Copy Orthologue
与Orthogroup Sequences目录相同的文件,但仅限于每个物种只包含一个基因的那些orthogroups。
 微信
微信 支付宝
支付宝






