系统发育基因组学(Phylogenomics)的介绍以及实操
Phylogenomics
写在前面
关于系统发育基因组学的内容,本文参考了 Mike Lee 的文章,将分为两期,本文介绍基本相关原理。
有一个相关的视频,时长为32’51’’ 感兴趣的可以点击下方进入观看。
什么是系统基因组学?
用一个容易理解但是不准确的概念来表示: 系统基因组学正试图在基因组水平上推断进化关系。
因为在实践中,我们并没有关注的所有生物体整个基因组。
并且根据我们考虑的多样性的广度,无论如何使用整个基因组是不可能或毫无意义的(因为它们可能太不同了)。
所以更恰当的说法是:系统基因组学试图在更接近基因组的水平上推断进化关系,而不是单个基因的系统发育(比如16S rRNA 基因树)。
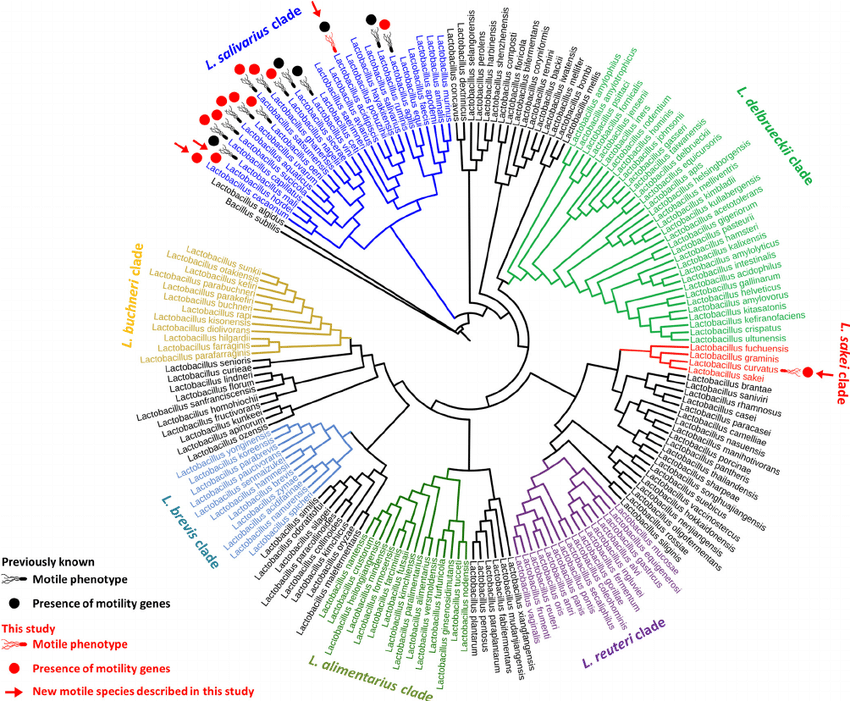
一般大家习惯于查看和使用的大多数系统发育树是单一基因类型(如 16S rRNA 基因)的不同拷贝去估计进化关系的视觉表示。
如果我们试图在生物体层面上思考,我们将使用这些基因拷贝作为代表生物体本身的代理,并且我们假设这些基因的进化关系告诉我们一些关于它们源生物进化关系的有意义的事情。
我们正在用系统基因组学做同样的事情,只是我们使用多个基因而不是单个基因。
最后,我们仍然只是使用序列作为代理。所以更合适的定义可能是:
系统基因组学试图推断由多个串联基因组成的序列之间的进化关系,同时假设这些推断的进化关系告诉我们一些关于其源基因组进化关系的有意义的信息。
什么是单拷贝核心基因?
单拷贝核心基因,或单拷贝基因 (single-copy genes SCGs),是我们目前正在谈论的大多数生物体中恰好存在 1 个拷贝的基因。
为了比较生物体之间的基因,我们需要考虑实际拥有这些基因的生物体。但是我们也需要单拷贝的基因(而不是在我们的目标生物体内往往存在多个拷贝的基因),因为系统发育学中的一个内置假设通常是所考虑的基因处于相似的进化压力下。
即使使用单拷贝基因,这也是微不足道的(并且可能永远不会真正完全正确),但是如果同一基因组中有多个基因拷贝(这些被称为旁系同源物),我们更有可能违反该假设。
所以这就是为什么当我们谈论系统基因组学时,SCG 扮演着如此重要的角色。

我们应该使用哪些基因?
我们选择研究的生物体决定了应该使用的基因。
根据上面谈到的关于为什么 SCG 在系统基因组学中如此有用,如果我们专注于更密切相关的生物群,那么符合这些标准的基因数量将比关注一个更分化的有机体组会相对较大。

上图来自 Hug et al. 2016 的生命之树使用了 15 个目标基因,因为它旨在跨越 3 个域。
但是,例如,如果我们只关注蓝藻,那么这一群体中共享的单拷贝基因的数量会大得多。
例如,为了为 GToTree 设计一个特定于蓝藻的单拷贝基因集,需要在 NCBI 提供的至少 90% 中蓝藻基因组中找到恰好存在于 1 个拷贝中的所有基因。
该过程在执行时产生了 251 个基因靶点。相比之下,当对所有细菌基因组应用相同的过程时,结果是 74 个。
没有一套理想的目标基因可以一直使用,因为它完全取决于我们试图研究的多样性的广度。
流程概览
假设有我们想要包含的基因组以及想要定位的一组单拷贝基因。下一步是什么?
目前最常见的方法之一可能如下所示:
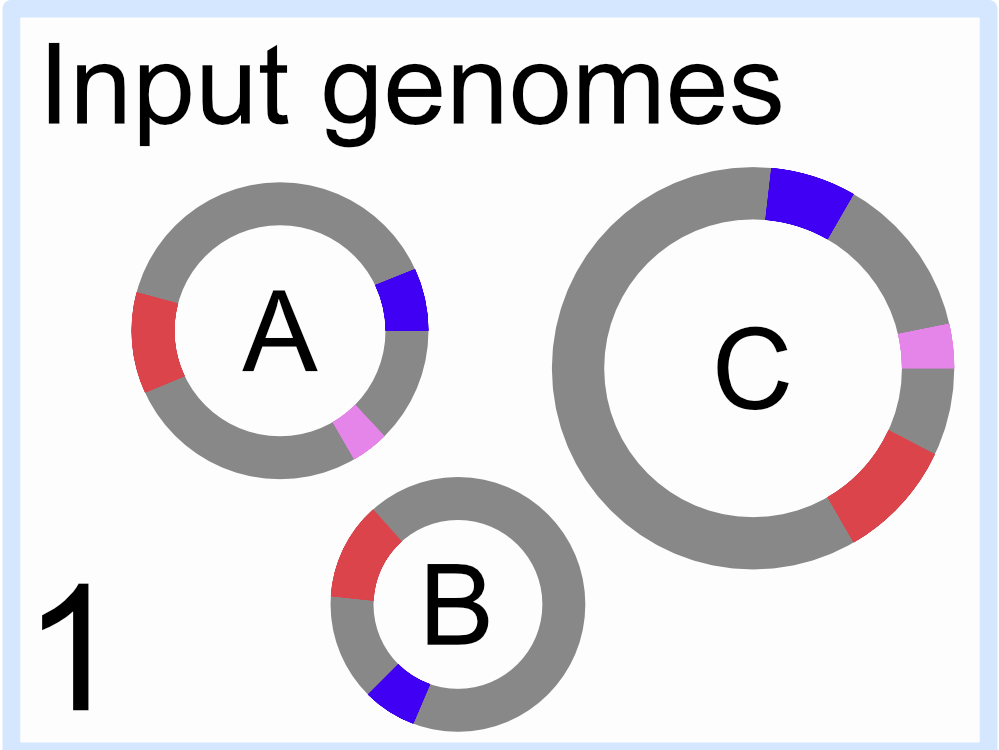
1. 在我们所有的输入基因组中识别我们的目标基因
- 这些由右侧卡通中的颜色表示
- 请注意,基因组 “B” 缺少 3 个示例目标基因之一
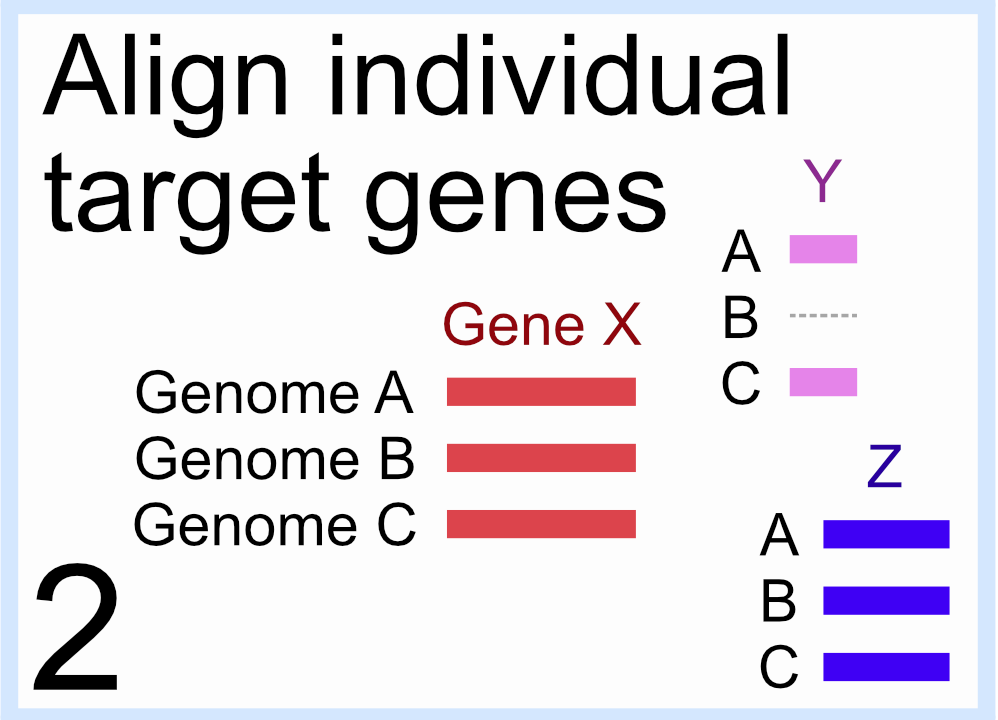
2. 单独比对每组识别的基因
- 例如 从所有输入基因组中识别出的目标基因“X”的所有拷贝都对齐在一起;对所有基因组单独完成相同的过程
- 对于缺少目标基因之一的任何基因组,将 gaps 插入到该基因组的该基因的比对中(如上图”Genome B”的基因“Y”的比对)

3. 将所有这些序列水平对齐排列
- 所有的单个基因比对并水平对齐排列,如果是氨基酸比对,通常会有一些“spacing”字符,比如几个“X”
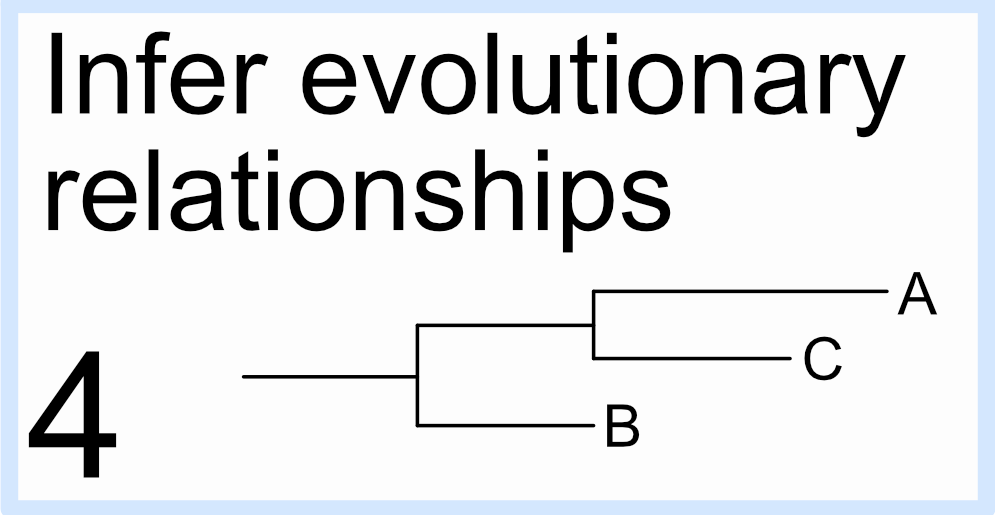
4. 推断这些组合序列的进化关系
- 构建进化树
这些一般步骤中的大多数可以以不同的方式完成,并且有很多决定需要做出,例如我们如何尝试识别我们的目标基因,我们使用什么来对齐它们,以及我们使用什么来制作我们的树。还有很多我们可能需要注意的事情,例如:
-
可能想要采取一些措施来进行某种形式的自动筛选,以确保我们不会让任何被确定为我们目标基因版本的基因通过,但可能不应该这样做(因为这可能会影响我们对该基因集的比对)。
-
如果一个给定的输入基因组中已识别出有目标基因的多个拷贝,我们可能不希望包含来自该基因组的该基因(具体可参见上面的SCG部分)。
-
如果输入基因组的目标基因总体上很少被识别,我们可能希望从分析中完全删除该基因组。
下面我们将使用 GToTree 做一些系统基因组学,我们将看到它如何为我们自动化这些过程。
系统基因组学实操
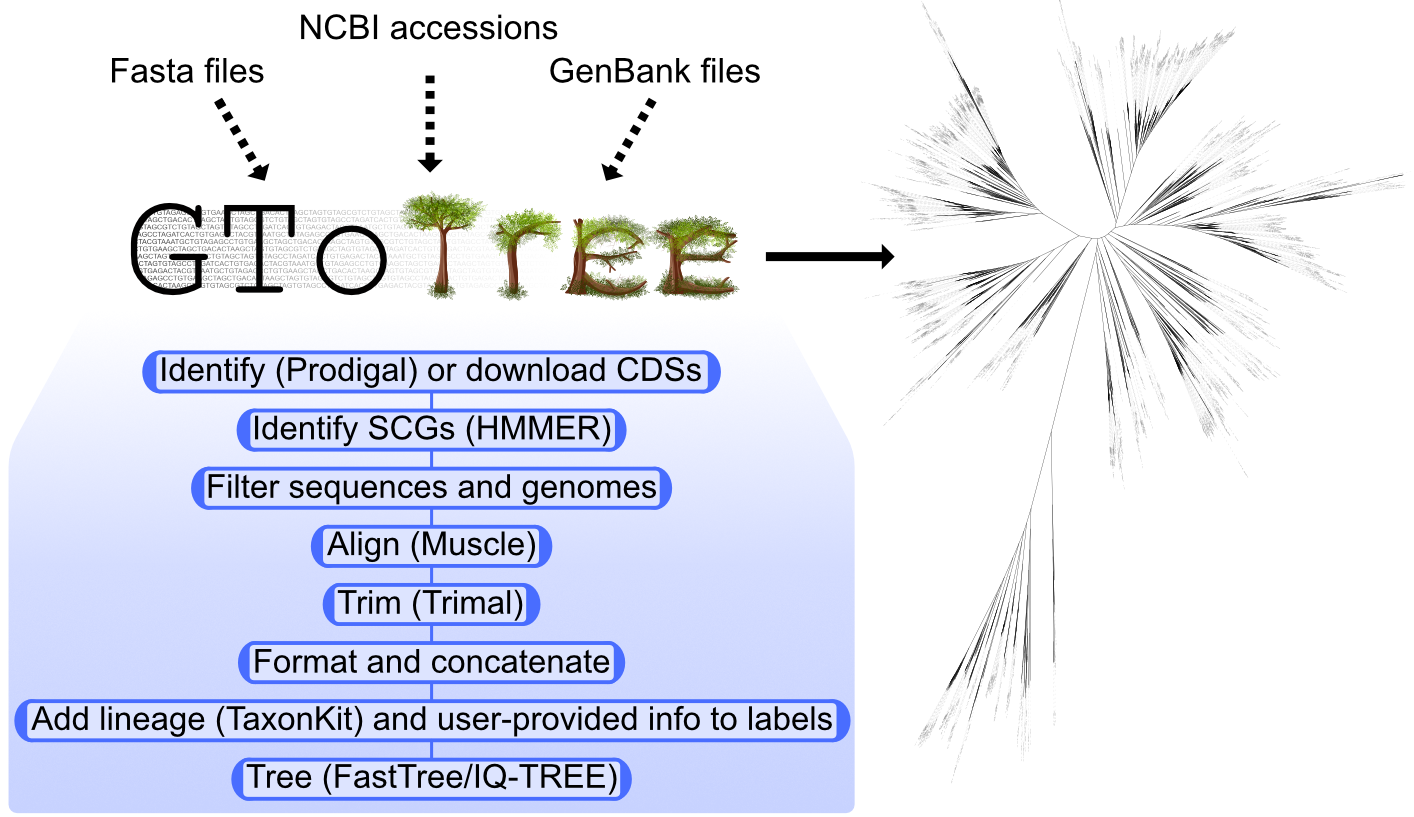
这里要用到一款关于软件 GToTree 这是一个系统基因组学工作流程的软件,具体的内容可以通过 Github 访问,当然这款软件也不是适用于所有的数据组,还需要去自己详细了解一下。
GToTree 是一个用户友好的系统基因组学工作流程。
它为我们处理了所有潜在的计算量大的任务,例如:
- 大规模访问基因组数据
- 整合来自不同文件格式的基因组
- 执行基因和基因组过滤
- 将 input labels 交换为 lineage 信息
- 为我们将流程中的不同工具拼接在一起;等等
它使系统发育树的生成和迭代更加容易处理。 GToTree wiki 上有更多信息,
这里展示其概览图:
安装 GToTree
使用 GToTree 创建一个 conda 环境,并像这样激活它:
1 | conda create -y -n gtotree -c conda-forge -c bioconda -c defaults -c astrobiomike gtotree |
聚球藻(Synechococcus)实例
聚球藻菌属(学名:Synechococcus)为聚球藻菌科的一个属。它是一种丰富的蓝藻,遍布全球海洋(和其他地方,但这里我们只讨论海洋 Synechococcus)
分类分配工具( taxonomy assignment tool )会告诉我们这些是聚球藻,这是必要的第一步
下一步是看看哪些聚球藻(Synechococcus)推断与新基因组最密切相关 —— 这就是从头系统发育树派上用场的地方(在使用新基因组时,无论是来自分离物测序或从宏基因组中恢复,一般在使用 GToTree 生成系统发育树之前使用GTDB-Tk 进行分类分配。)
我们需要提供给 GToTree 两件必需的内容:
- 我们想要研究哪些基因组;
- 我们想用哪些目标基因来构建我们的系统发育树。
输入基因组
把新基因组(FASTA格式)放入一些海洋参考聚球藻中(只需要提供 NCBI accessions 之后 GToTree 会为我们获取它们)。
我们可以使用以下内容下载此起始示例数据:
1 | curl -L -o syn-gtotree-example.tar.gz https://ndownloader.figshare.com/files/23629763 |
如果我们查看该ref-syn-accs.txt文件,我们可以看到其中有 20 个 NCBI 程序集:
1 | wc -l ref-syn-accs.txt |
1 | GCF_000011385 |
其中包括 19 种海洋聚球藻参考,以及 Gloeobacter violaceus(一种更远的蓝藻)作为外群创建有根树🌲。
注意
要生成此参考 accessions,我们可以在 NCBI 网站的 assembly 区域中进行搜索,也可以使用 EDirect 从命令行搜索它们。在此处的 GToTree wiki Alteromonas example 中,有两种方式生成我们的参考 accessions 的示例
我们可以看到我们有 12 个新的基因组序列作为 fasta 文件:
1 | ls *.fa | wc -l |
我们只需要将它们放入一个可以提供给 GToTree 的文件中,例如:
1 | ls *.fa > our-genome-fasta-files.txt |
1 | GEYO.fa |
指定我们的目标基因
在这种情况下,我们正在处理所有聚球藻,它们是蓝藻目( Cyanobacteria )。
我们可以通过GToTree的 gtt-hmms 看到 HMM 目标基因集:
1 | gtt-hmms |
它告诉我们 HMM 的存储位置(也可以根据需要自定义),然后列出当前可用的内容:
1 | The environment variable GToTree_HMM_dir is set to: |
有一组蓝藻的 HMM 目标基因集,其中包含 251 个基因,适用于对聚球藻的关注。
GToTree 中包含的 SCG-sets 是按此处所述生成的。
运行 GToTree
GToTree 的帮助菜单可以通过 GToTree -h 看到
在这里,我们将启动命令,然后分解代码以及 GToTree 在运行时所做的事情。
在这里运行时,迄今为止花费的最大时间是进行比对(使用更多线程运行意味着可以同时运行更多比对,从而大大提高运行时)。
通过比对和制作进化树,蓝藻 HMM 集中的全部 251 个基因,这在笔记本电脑上花费大约 6 分钟。
如果想跳过处理,下面会提供结果文件。
1 | GToTree -a ref-syn-accs.txt -f our-genome-fasta-files.txt -H Cyanobacteria -t -L Species -j 4 -o Syn-GToTree-out |
参数详解
1 | GToTree |
当我们执行该命令时,在此示例中为 GToTree:
- 下载提供的每个参考的氨基酸序列
- 对所有输入的fasta文件调用开放阅读框架(使用 prodigal )
- 识别目标基因(使用 HMMER3 )
- 根据目标基因估计完成度/冗余度
- 根据长度和基因组根据目标基因的命中数过滤掉 gene-hits
- 为缺失任何基因的基因组添加所需的 gap 序列
- 将每个单独的基因集使用 Muscle 进行比对
- 使用 Trimal 执行比对的自动修剪
- 将所有比对( alignments )连接在一起
- 默认使用 FastTree 建树
制作 iToL 映射文件来为新基因组着色
这一步是可选的,在这里要做的最后一件事是快速制作颜色映射文件,以便在 Interactive Tree of Life (iToL)站点上显示新基因组,我们将在其中可视化我们的树。
这需要一个输入文件,该文件只包含我们想要着色的树上的标签。
在这里,可以从我们的输入 FASTA 列表文件删除后缀 “.fa” 快速制作:
1 | $ cat our-genome-fasta-files.txt |
1 | cut -f 1 -d "." our-genome-fasta-files.txt > labels-to-color.txt |
现在我们可以使用 GToTree 帮助程序来快速制作 iToL 兼容文件:
1 | gtt-gen-itol-map -g labels-to-color.txt -o iToL-colors.txt |
可视化
我们需要在本地计算机上生成的输出文件是Syn-GtoTree-out.tre文件和iToL-colors.txt 文件,以便能够将它们上传到 iToL 网站。
可以使用左侧的文件导航器面板从活页夹环境下载这些文件,或者可以通过单击以下链接下载这两个文件:
我们可以上传我们的树以在 iToL 网站上对其进行可视化:
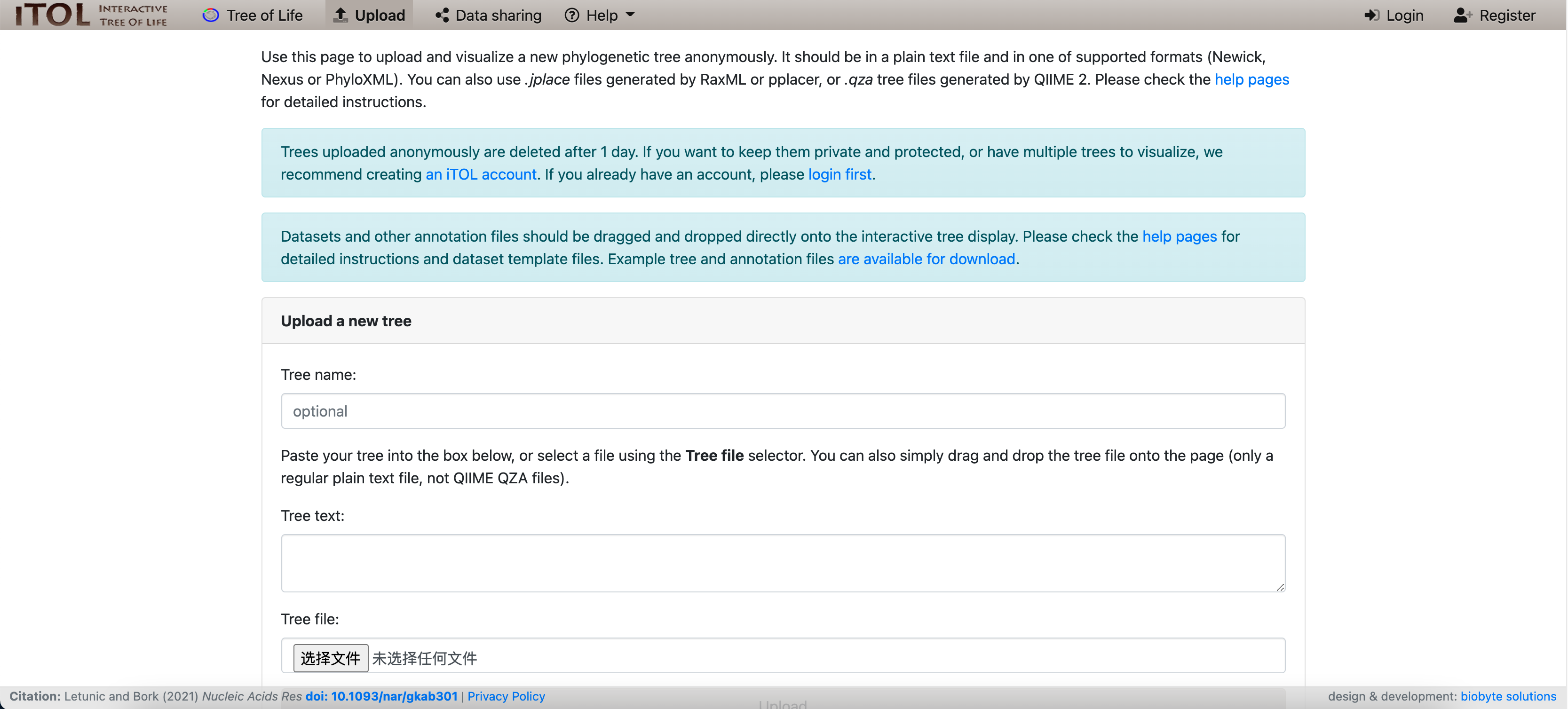
我们可以将 Syn-GtoTree-out.tre 文件拖放到:
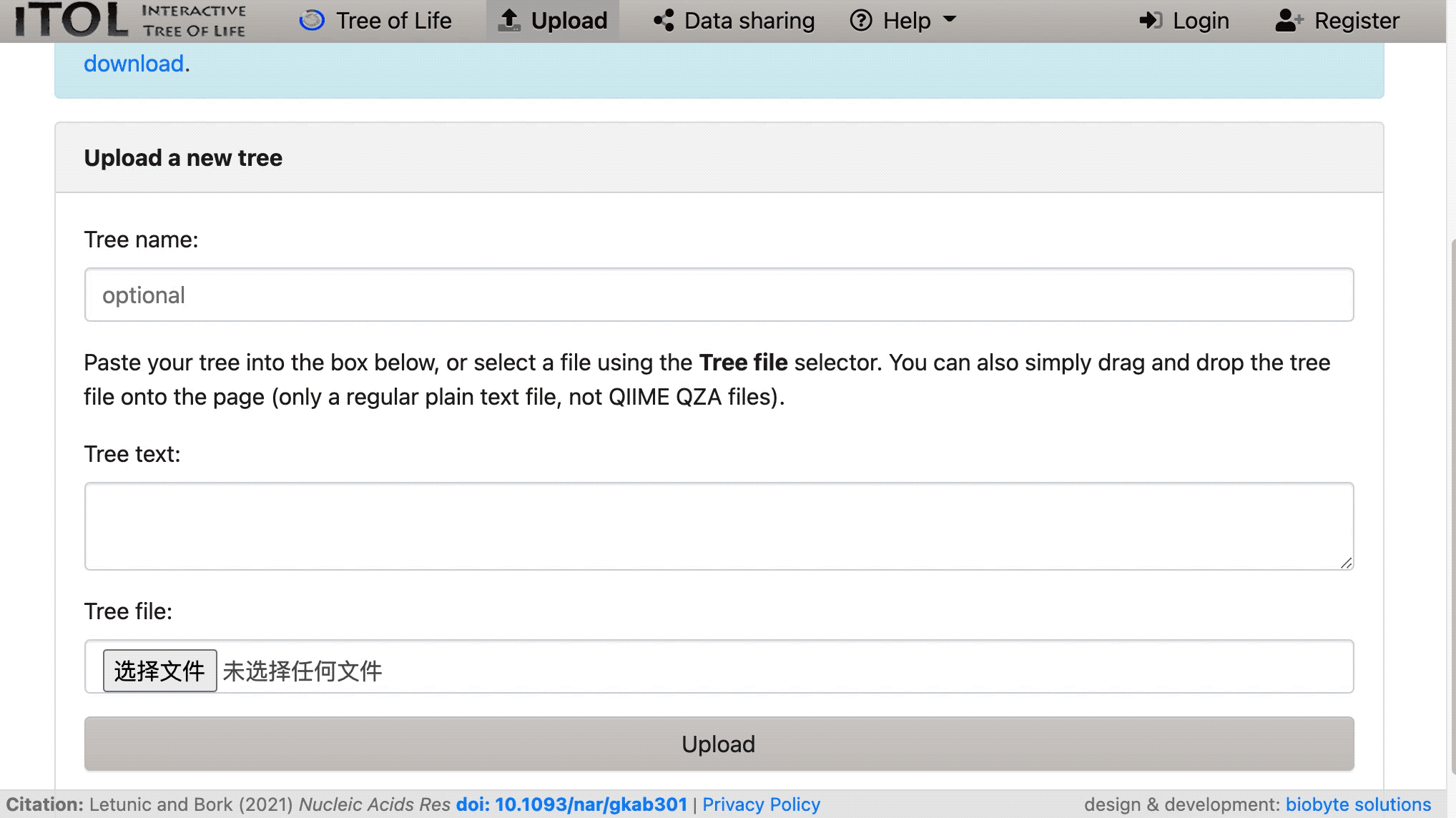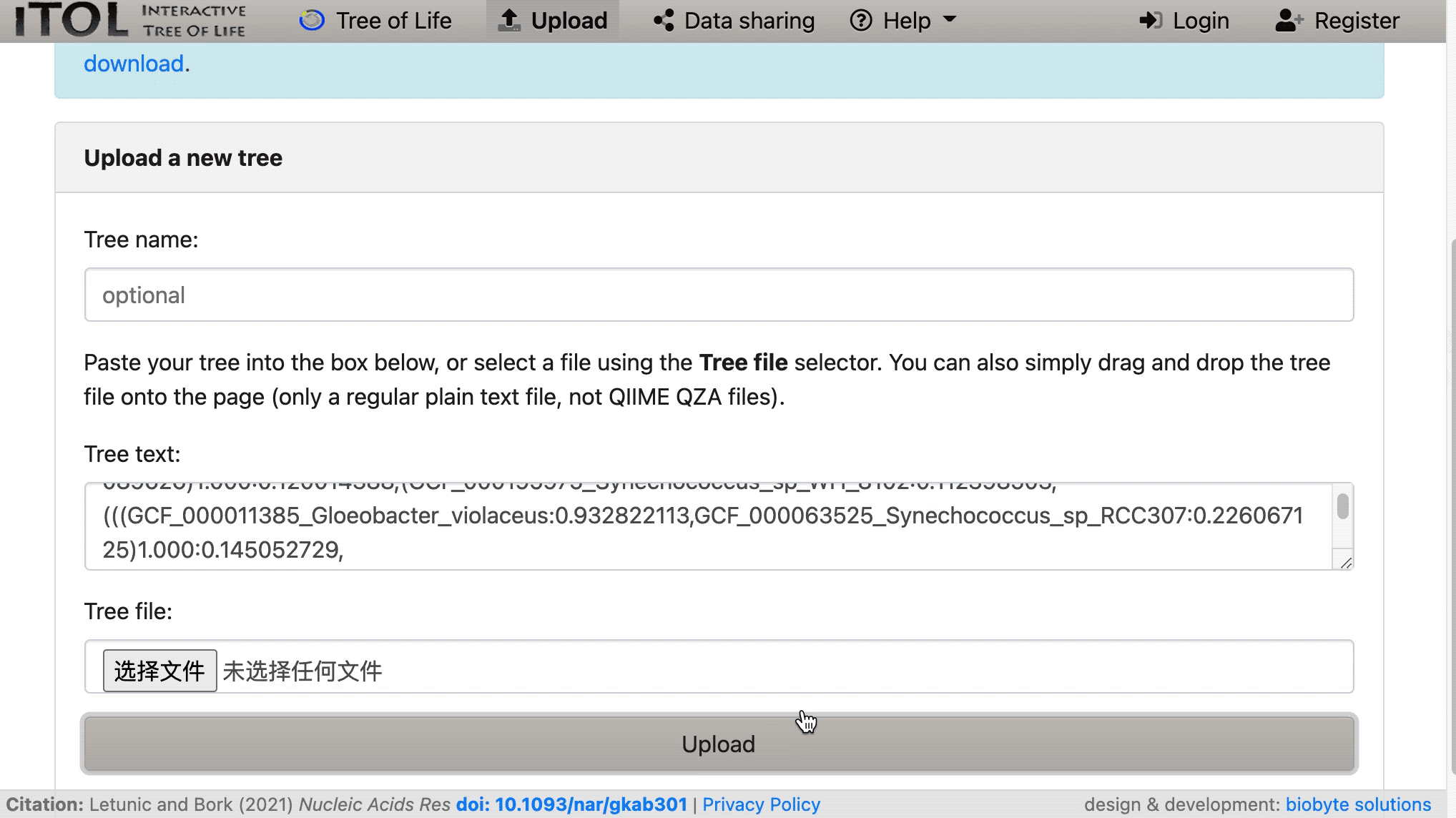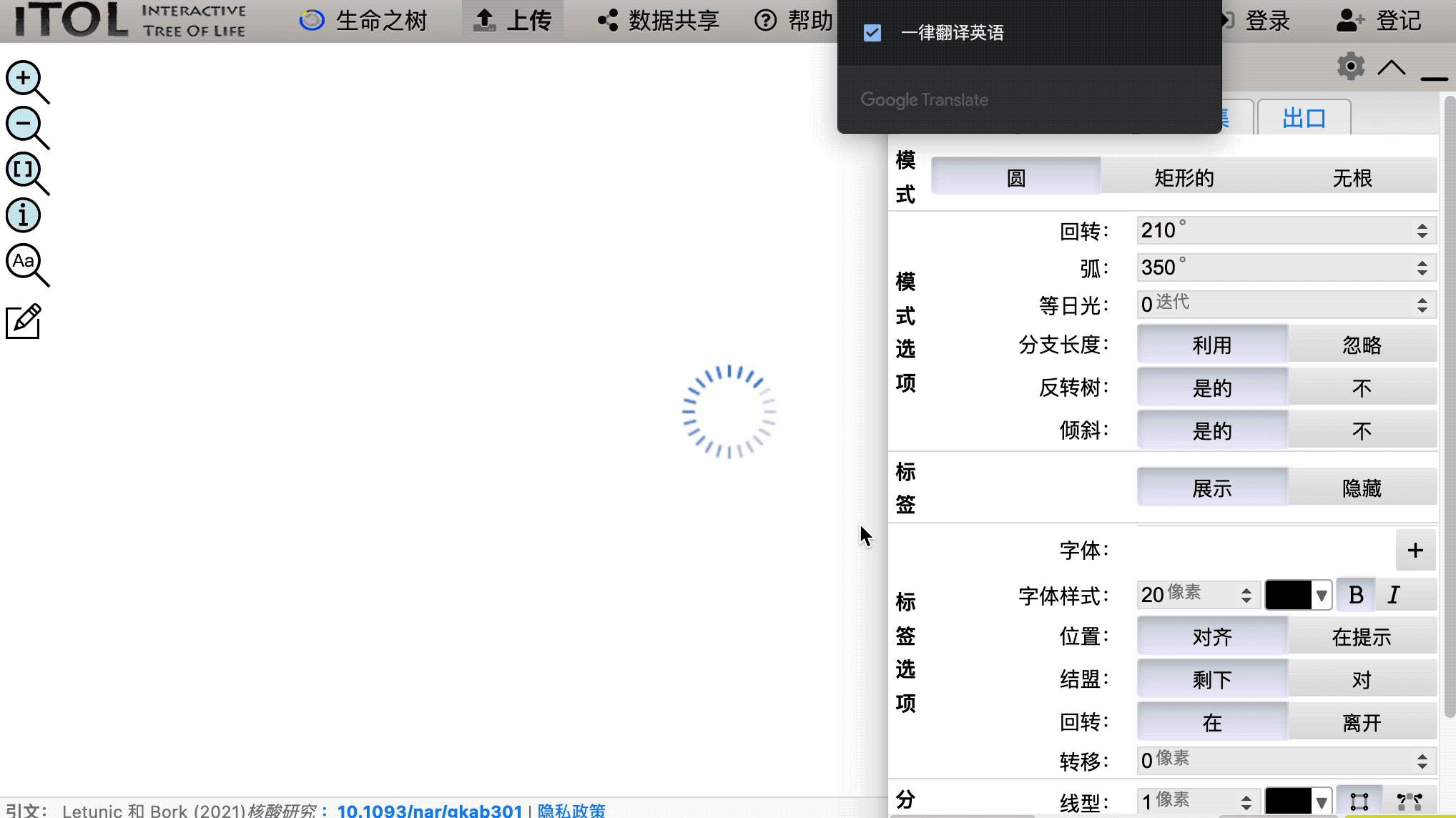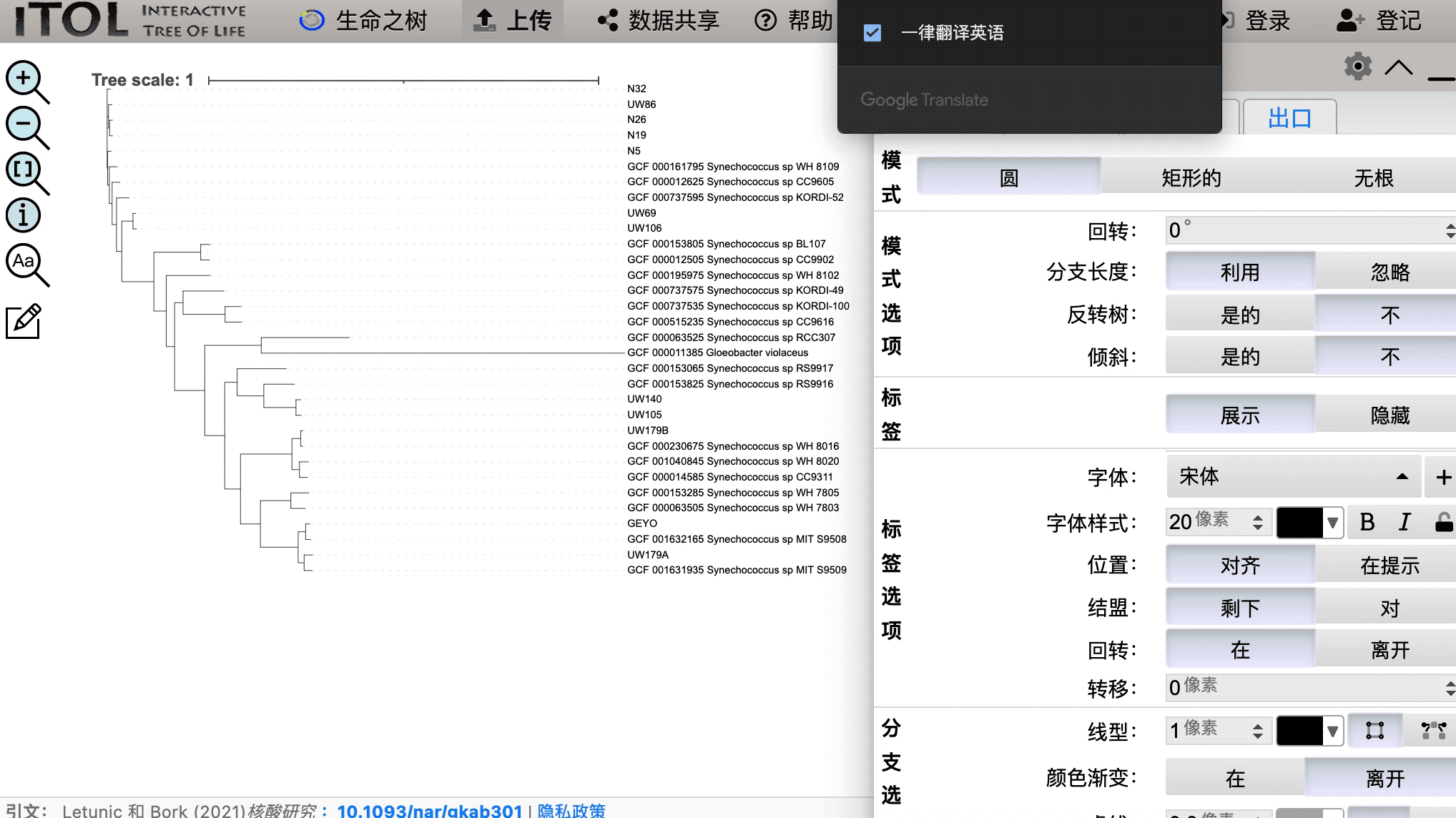
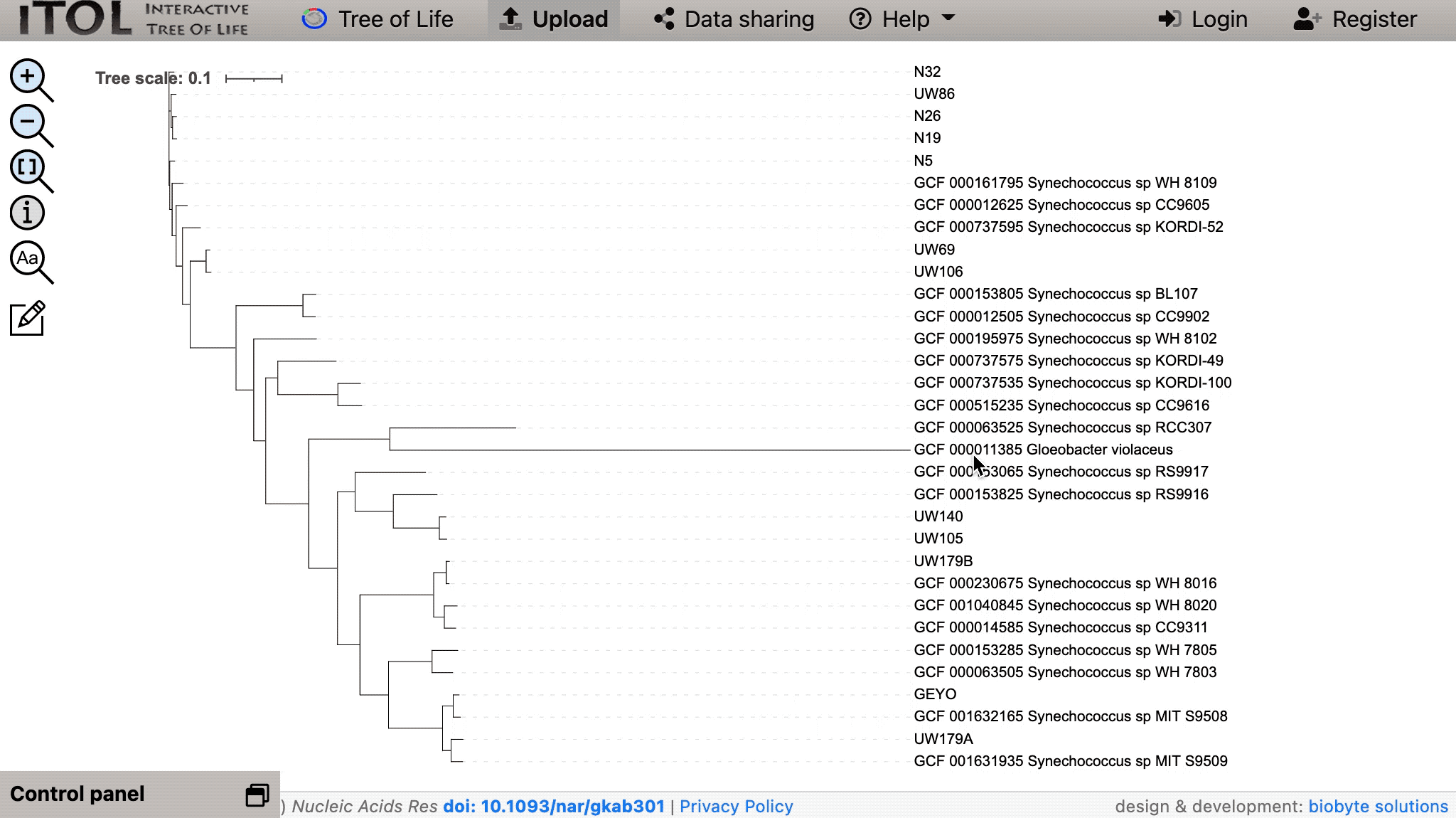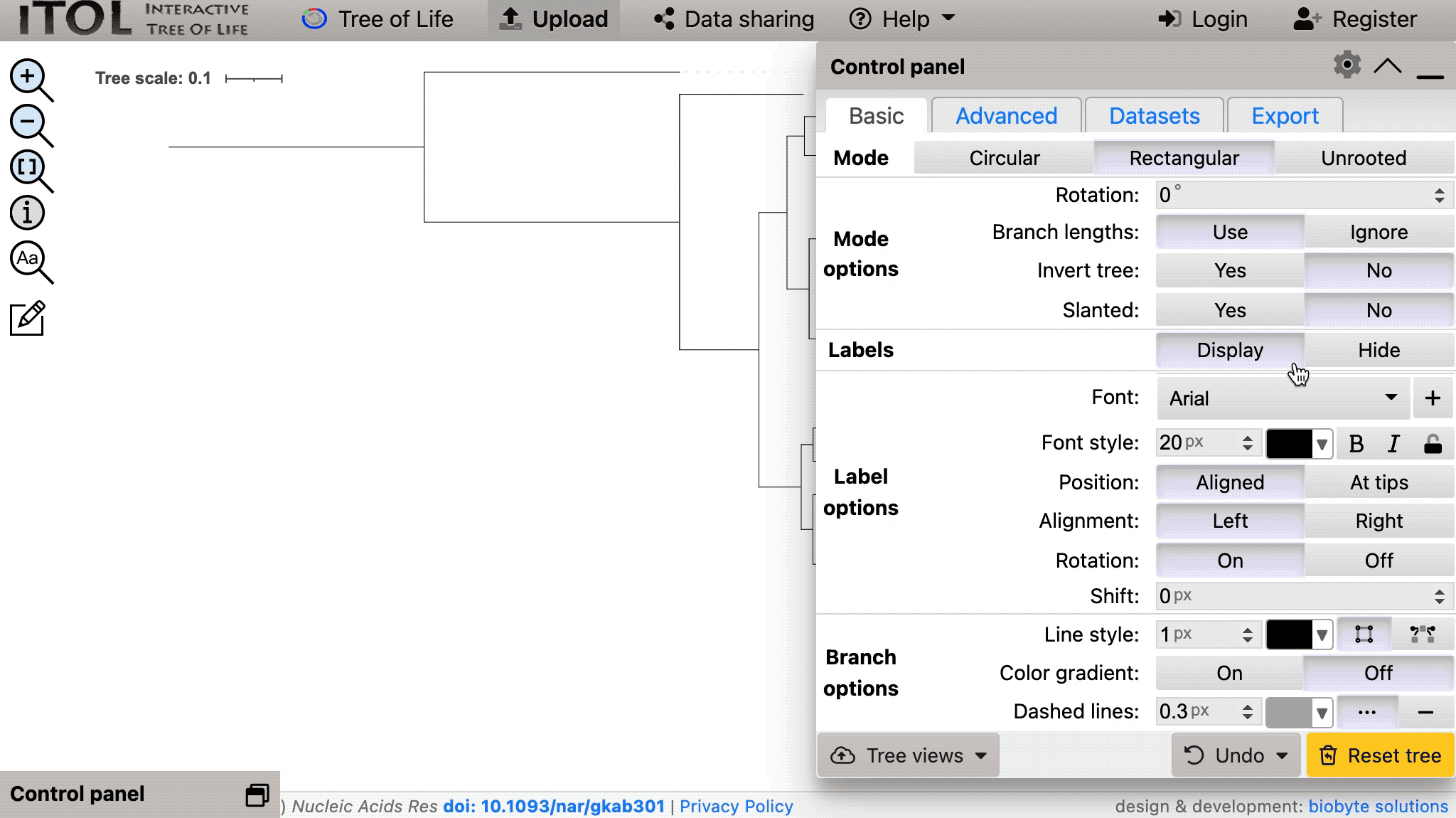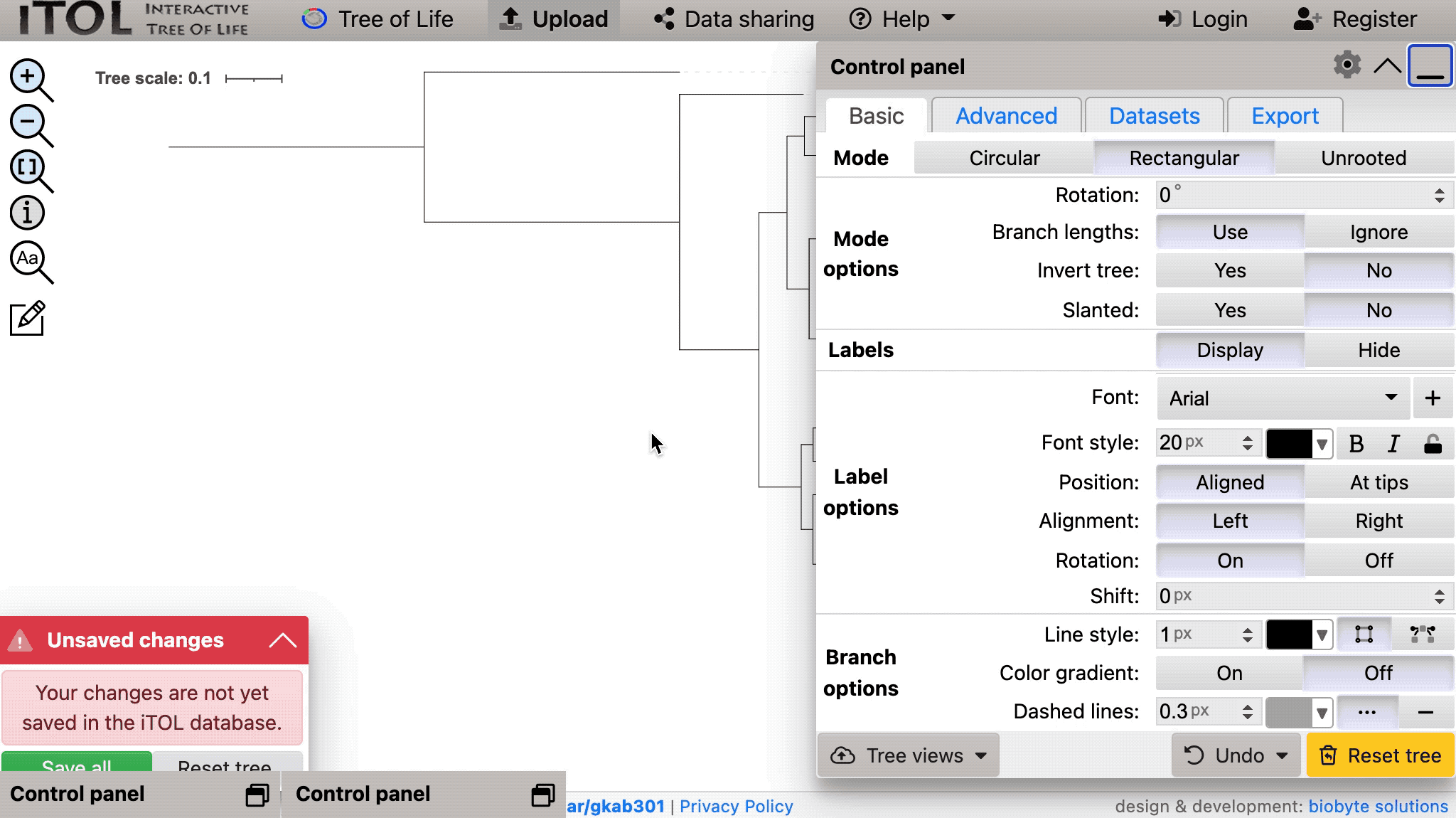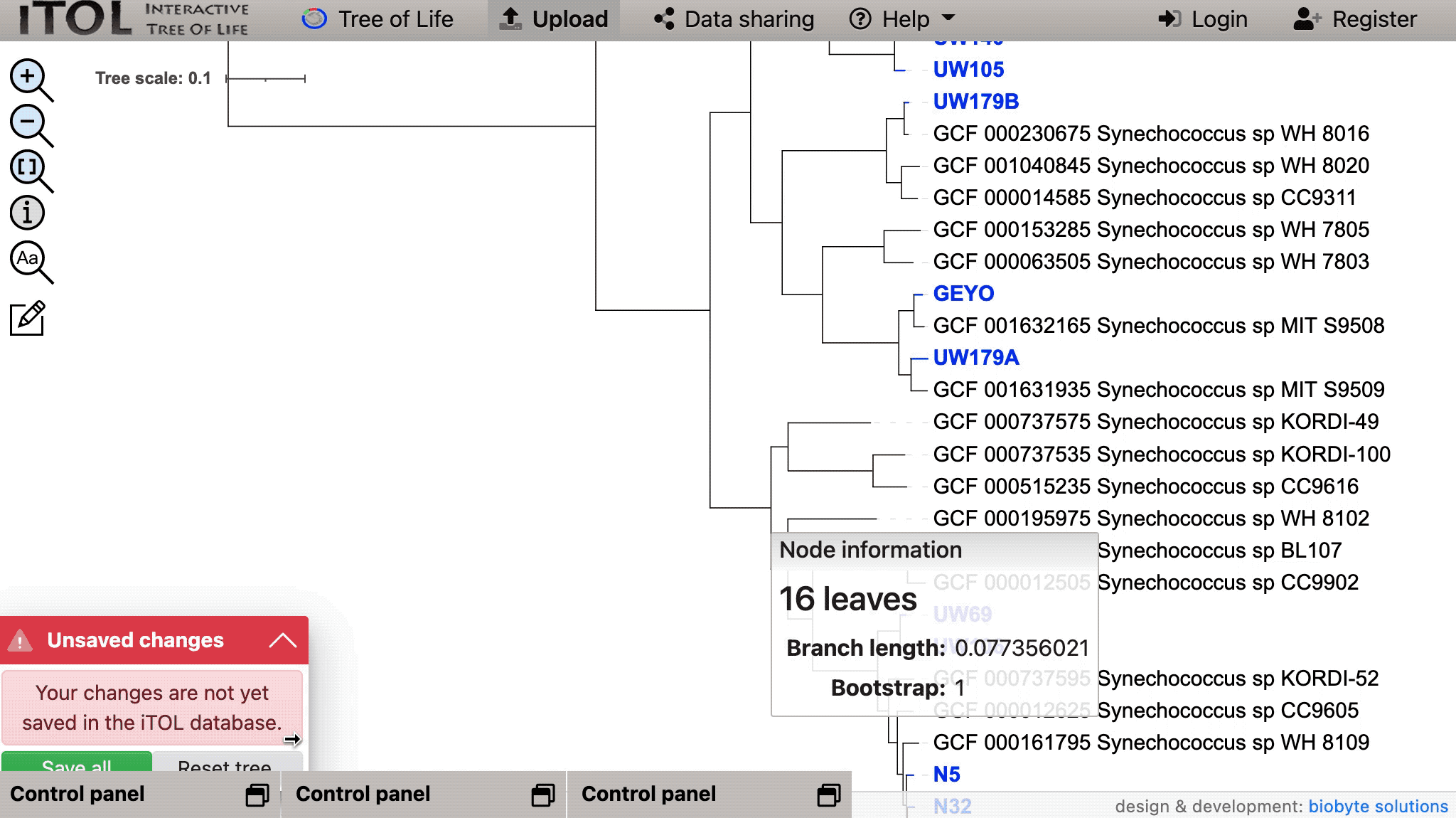
然后将加载我们的树:
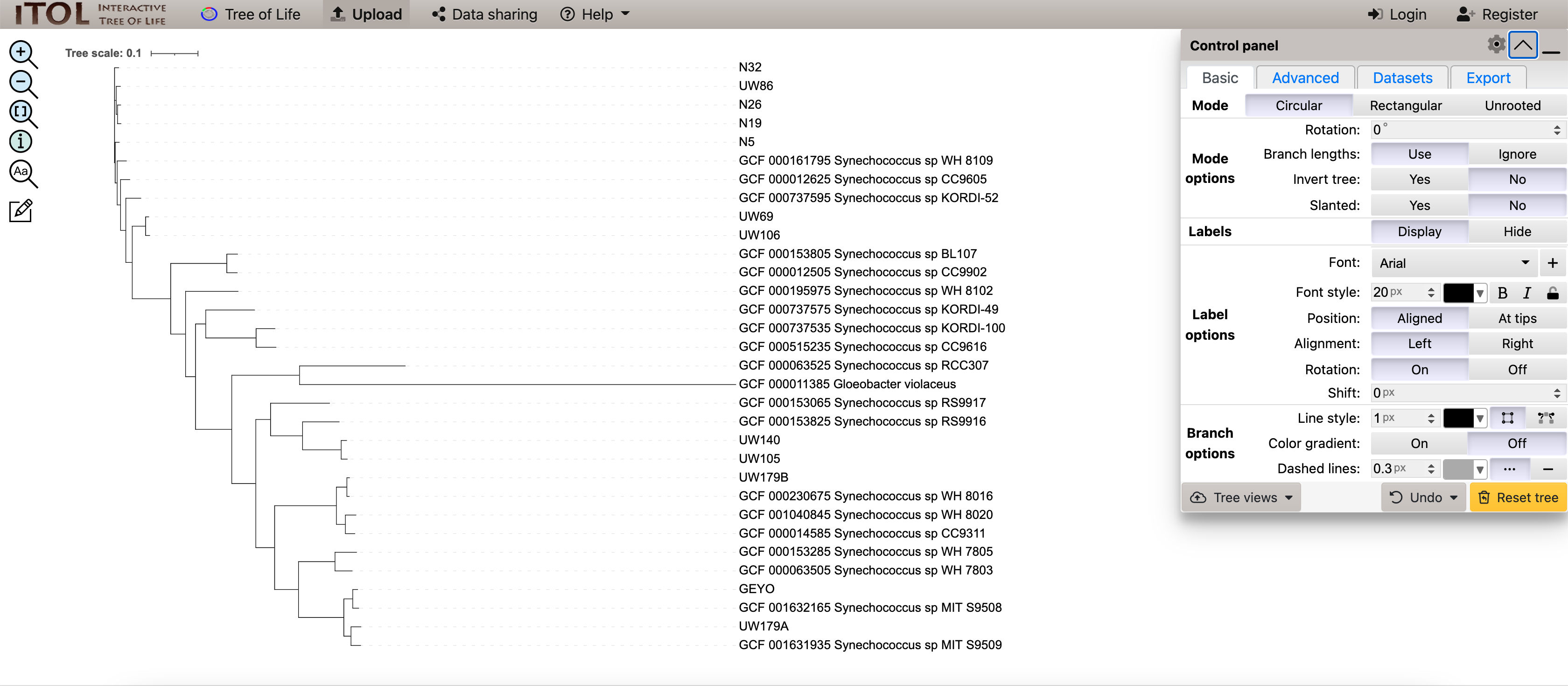
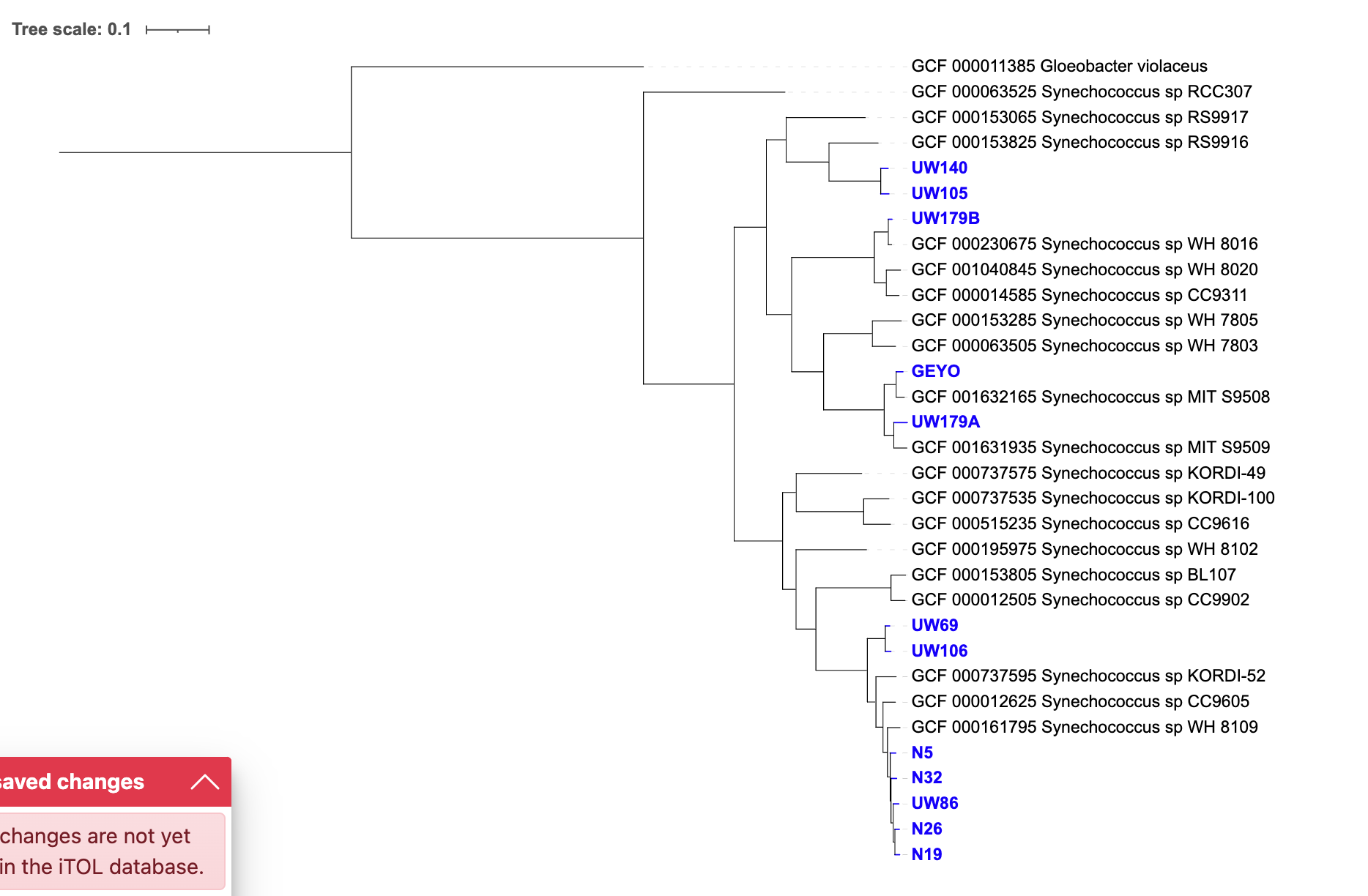
现在我们可以将我们的树植根于 Gloeobacter violaceus:
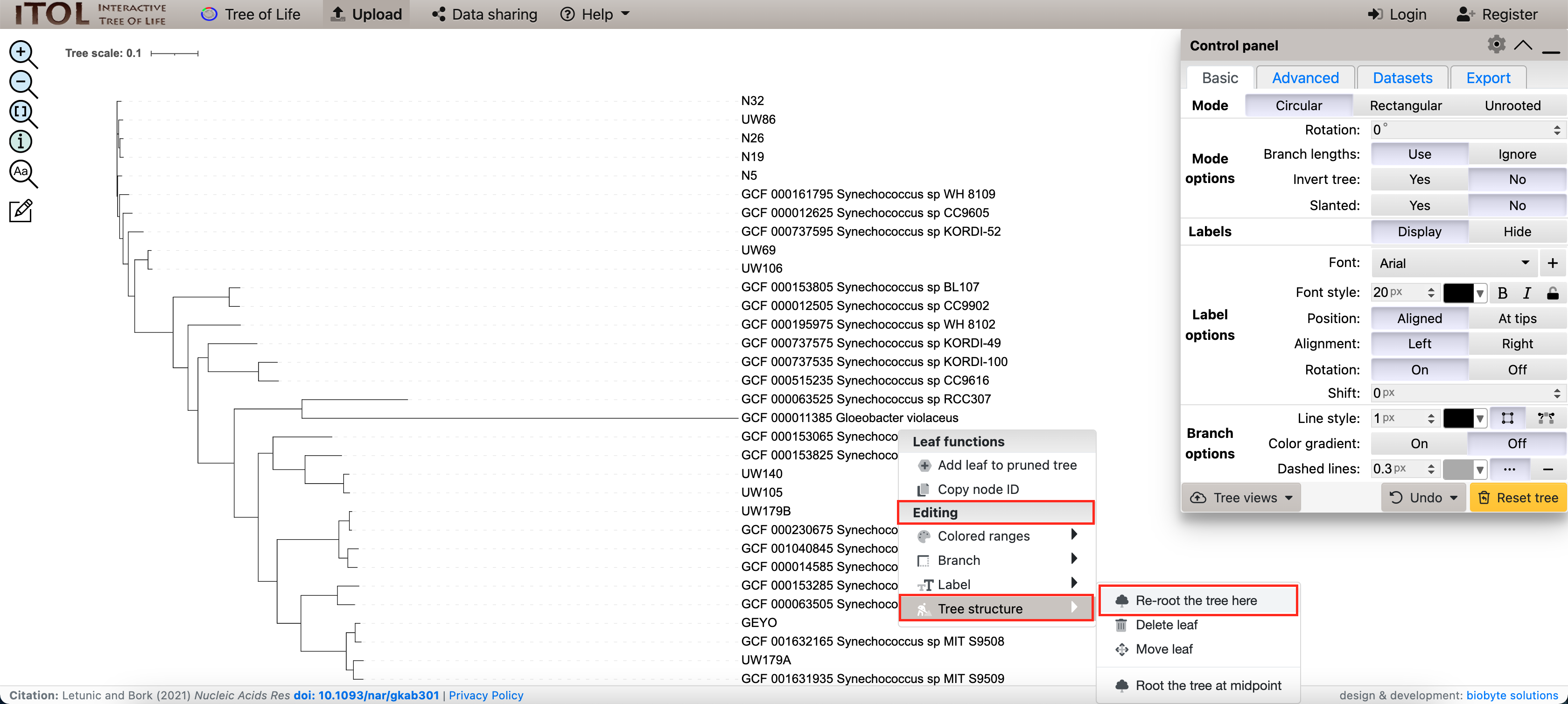
最后,我们可以将 iToL-colors.txt 文件拖放到树上以突出显示我们的新基因组:
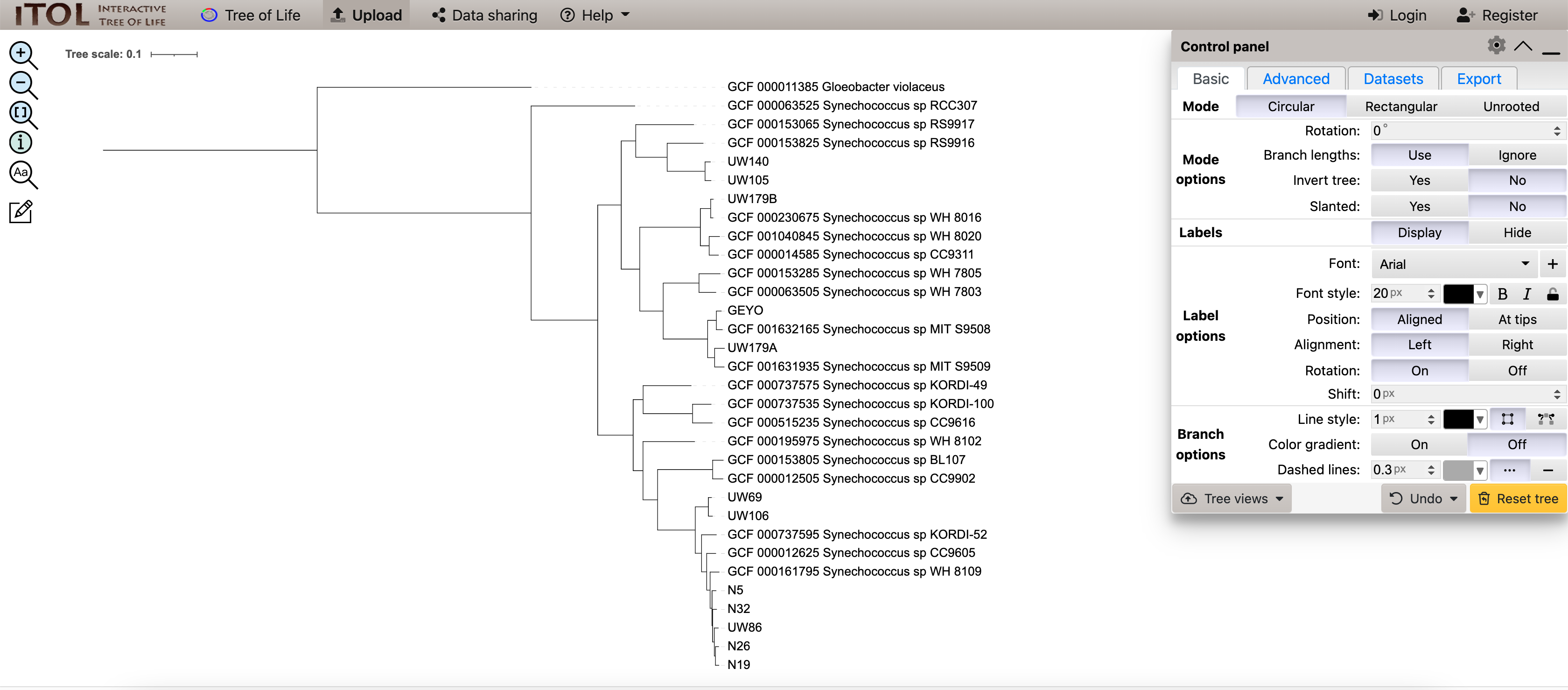
最终结果得出这样的树状图:
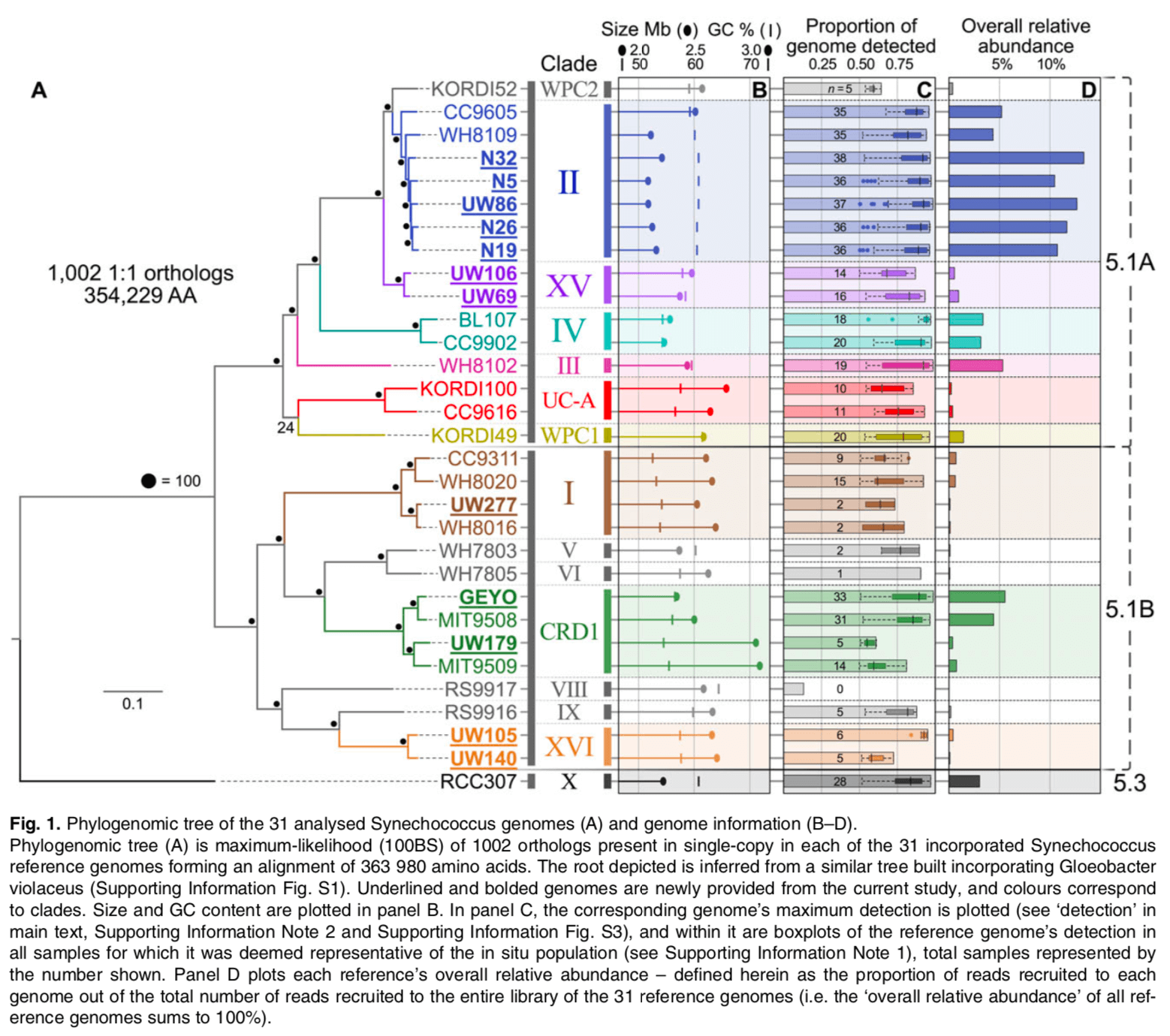
一般来说,对于给定的一组感兴趣的生物,最好使用与该组生物一样多的单拷贝核心基因。
但是,为了获得满足我们当前需求的可信信号,这并不总是必要的。
在这里,蓝藻特异性 SCG-set 的 251 个基因给出了与其论文图 1 中的 1,002 个目标基因树中获得的几乎相同的树和关系(除了一些旋转、着色和标签调整):
其他有用的输出
除了树文件之外,GToTree 还生成了几个文件。
- 比对文件(我们可以使用不同的建树软件制作我们的树)
- 一个基因组汇总表,其中包含每个基因组的信息,例如估计的完成/冗余百分比、谱系信息、输入标签和树中的最终标签等。
- 单拷贝基因命中表,每个基因组每个目标基因的单拷贝基因命中计数
- 运行日志
- 一个分区文件,可以使用混合模型制作树(示例在下面的更多示例下链接)
- 引用文件,列出所有使用的程序和相关参考文献
参考
Happy Belly Bioinformatics: an open-source resource dedicated to helping biologists utilize bioinformatics
Lee M D. GToTree: a user-friendly workflow for phylogenomics[J]. Bioinformatics, 2019, 35(20): 4162-4164.
Lee M D, Ahlgren N A, Kling J D, et al. Marine Synechococcus isolates representing globally abundant genomic lineages demonstrate a unique evolutionary path of genome reduction without a decrease in GC content[J]. Environmental microbiology, 2019, 21(5): 1677-1686.
 微信
微信 支付宝
支付宝





