ALLHIC使用 | HiC辅助基因组组装(三)
安装
1 | git clone https://github.com/tangerzhang/ALLHiC |
依赖软件
- samtools v1.9+
- bedtools
- matplotlib v2.0+
写在前面
官网链接手册整体流程
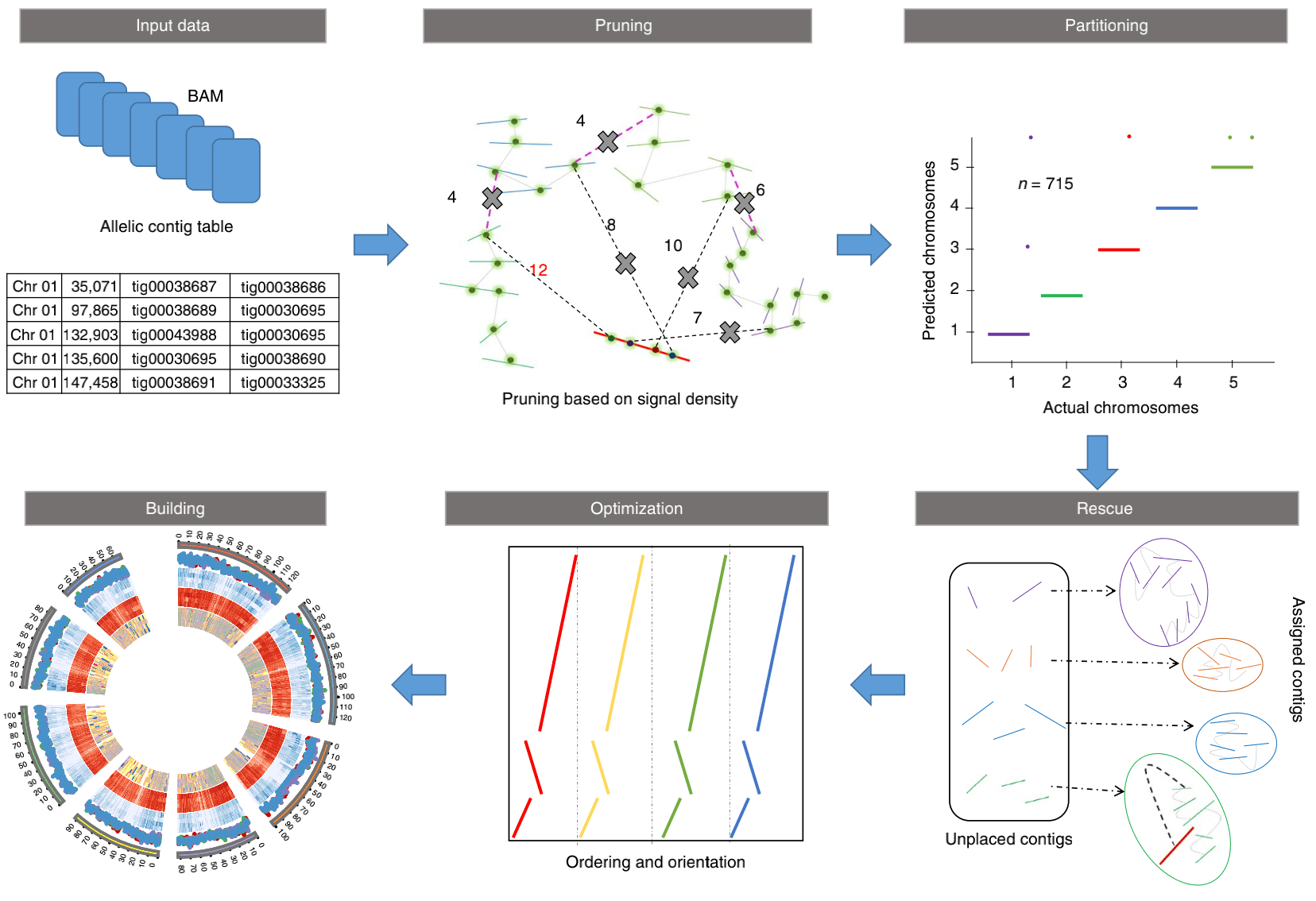
ALLHiC一共分为五步:pruning, partition, rescue, optimization, building
-
prune 步骤去除了等位基因之间的联系,因此同源染色体更易于单独分离。
-
partition 功能将修剪的bam文件作为输入,并根据Hi-C建议的链接对链接的contigs进行聚类,大概是沿着相同同源染色体在预设数量的分区中进行。
-
rescue 功能从原始未修剪的bam文件中搜索分区步骤中不涉及的contigs,并根据Hi-C信号密度将它们分配给特定的群集。
-
optimize 步骤采用每个分区,并优化所有contigs的顺序和方向。
-
build 步骤通过连接contigs来重建每个染色体
如下图所示:
 ]
]
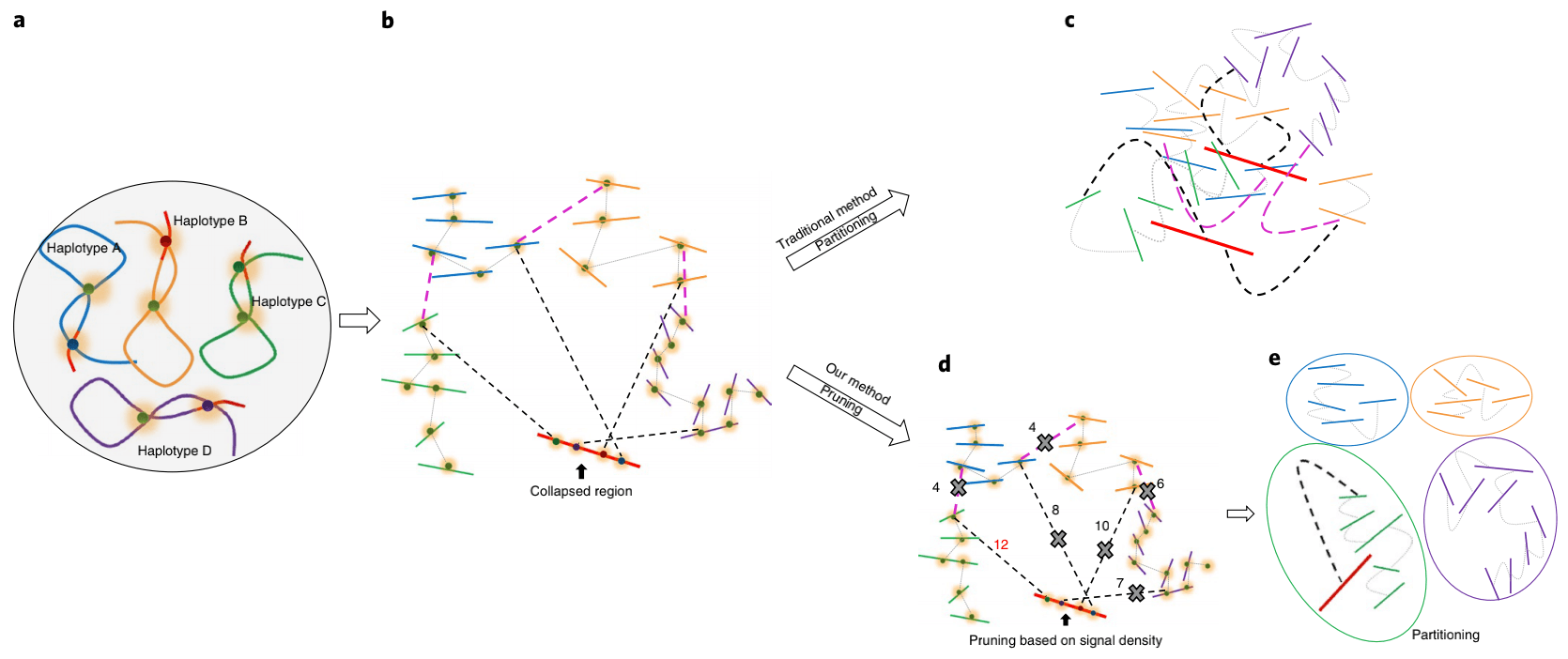
Explanation of Prune
-
同源四倍体基因组的示意图。四个同源染色体显示为不同的颜色(分别为蓝色,橙色,绿色和紫色)。染色体中的红色区域表示具有高度相似性的序列。
-
检测自身四倍体基因组中的Hi-C信号。黑色虚线表示折叠区域和未折叠区域contigs之间的Hi-C信号。粉色虚线表示单体型Hi-C链接,灰色虚线表示单体型Hi-C链接。在组装过程中,红色区域会因高度的序列相似性而崩溃;同时,如果其他区域之间存在大量差异,则会将它们分为不同的contigs。由于塌陷区域与来自不同单倍型的contigs在物理上相关,因此将在塌陷区域与所有其他未塌陷的contigs之间检测到Hi-C信号。
-
传统的Hi-C脚手架方法将检测来自不同单倍型和折叠区域的contigs中的信号,并将所有序列聚在一起。
-
修剪Hi-C信号:1-去除等位基因区域之间的信号;2-仅在折叠区域和未折叠contigs之间保留最强的信号。
-
基于修剪的Hi-C信息进行分区。理想情况下,根据修剪结果将contigs分为不同的组。
运行ALLHIC
数据准备
- Contig 水平的基因组组装结果
- Hi-C测序数据
将 Hi-C reads Map 到基因组草图
bwa index and samtools faidx 对基因组草图建立索引
1 | bwa index -a bwtsw draft.asm.fasta |
Hi-C reads Aligning 到基因组草图上
1 | bwa aln -t 24 draft.asm.fasta reads_R1.fastq.gz > sample_R1.sai |
过滤SAM文件
1 | PreprocessSAMs.pl sample.bwa_aln.sam draft.asm.fasta MBOI |
Prune(option)
1 | Usage: |
在这里需要输入Allele.ctg.table文件,这个文件需要自己获取,参考官方参考链接;
方法一:基于 BLAST 结果鉴定等位基因重叠群
1 | $ blastn -query rice.cds -db Bd.cds -out rice_vs_Sb.blast.out -evalue 0.001 -outfmt 6 -num_threads 4 -num_alignments 1 |
移除同一性 < 60% 和覆盖率 < 80% 的blast hits
1 | blastn_parse.pl -i rice_vs_Sb.blast.out -o Erice_vs_Sb.blast.out -q rice.cds-b 1 -c 0.6 -d 0.8 |
根据 BLAST 结果对等位基因进行分类
1 | classify.pl -i Eblast.out -p 2 -r Sbicolor_313_v3.1.gene -g rice.gff3 |
方法二:基于 GMAP 的方法
运行 gmap 得到一个 gff3 文件
1 | gmap_build -D . -d DB target.genome |
perl gmap2AlleleTable.pl referenece.gff3
gmap2Alleletable.pl的脚本链接点击此处
gmap2Alleletable.pl内容如下
1 | #!/usr/bin/perl -w |
Partition
ps :如果跳过了第四步,那么可以直接用第三步分析的结果sample.clean.bam
1 | Usage: |
Rescue
1 | #如果做了第四步,会生成 -c prunning.clusters.txt 以及 -i prunning.counts_AAGCTT.txt |
Optimize
1 | # 生成.clm文件 |
Build
1 | ALLHiC_build draft.asm.fasta |
plot
1 | ALLHiC_plot sample.clean.bam groups.agp chrn.list 500k pdf |
作者问题回复
-
修剪步骤中折叠区域和嵌合重叠群之间有什么区别?
在多倍体基因组中,一些同源区域(即等位基因序列)高度相似。这些序列经常被组装成一个重叠群,因为组装者不能分离等位基因。这种区域是折叠区域。
另一方面,一些重叠群包含来自不同单倍型或非同源染色体的序列,可以将其视为chimeric contigs。ALLHiC 旨在最大限度地减少collapsed contigs的负面影响,我们的模拟数据显示 ALLHiC 能够tolerate ~20% 的collapsed contigs;然而,只有约 5% 的chimeric contigs.。 -
如何确定哪些染色体组是同源的?
 微信
微信 支付宝
支付宝











