使用同源建模预测蛋白质结构
什么是蛋白质?
蛋白质是大的生物分子,负责执行生物体细胞内的大部分功能,包括对刺激作出反应、作为其他反应的催化剂、将分子从一个地方运输到另一个地方以及执行细胞信号传导。就像 DNA 序列一样,蛋白质序列是一串分子,但与 DNA 序列不同的是,有20种不同的称为氨基酸的分子构成了蛋白质序列。
蛋白质结构
每个1D 蛋白质序列串都折叠成3D 结构。这些 3D 蛋白质结构决定了蛋白质如何响应各种环境以及它与哪些其他分子相互作用,因此对于蛋白质执行其功能的能力至关重要。蛋白质的 3D 结构是通过提供蛋白质中每个原子在3D 空间中的坐标 (xyz)来描述的。
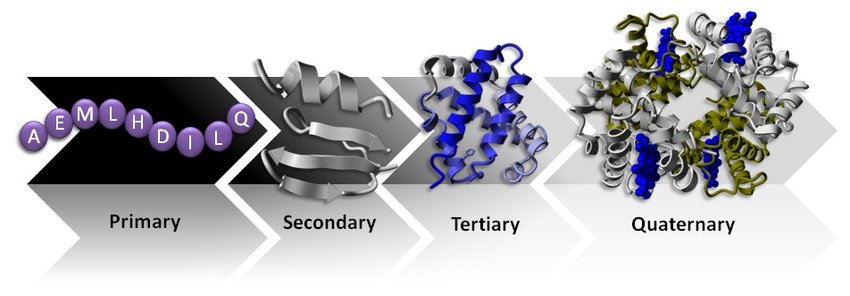
确定蛋白质结构
可以使用X 射线晶体学和核磁共振 (NMR)等实验程序确定蛋白质结构。
然而,这些技术缓慢且繁琐,并且不能应用于所有蛋白质。因此,高通量计算方法用于从序列预测蛋白质的 3D 结构。
同源建模
蛋白质结构预测最流行的计算方法之一是同源建模。
同源建模利用蛋白质结构的进化保守性来预测蛋白质的 3D 结构。从相同的共同祖先(同源性)进化而来的两种蛋白质往往具有相似的 3D 结构。
在同源建模中,这种蛋白质结构保守性的特性用于预测新发现的蛋白质序列的结构,这些蛋白质序列的结构无法使用传统的实验方法解析。
其主要思想是根据蛋白质序列数据库搜索未知结构的蛋白质序列,其中所有蛋白质的结构在实验上都是已知的,未知结构是根据数据库中进化上最接近或最匹配的蛋白质建模的。
在这篇文章中,我们描述了同源建模的方法,即它是如何工作的。我们还描述了如何使用 SWISS-MODEL 工具进行同源建模。
同源建模方法详解
在本节中,我们将概述同源建模所涉及的步骤。请注意,其中许多步骤是活跃的研究领域。
前面提到过,同源性建模始于对许多蛋白质的结构及其序列的了解,这些结构已通过实验方法确定。该方法使用这些先前的知识来预测我们知道序列但还不知道 3D 结构的蛋白质的结构。
为了预测蛋白质的结构,我们将首先预测 N、Ca、Cb(骨架)的坐标,然后是每个氨基酸的 R 基团(侧链)的坐标。
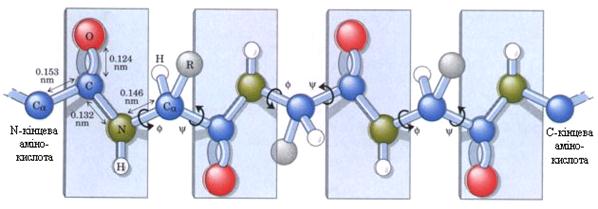
(一) 模板识别和初始对齐
首先,我们找到进化上最接近目标的蛋白质(我们希望预测其结构的蛋白质)。
这是使用数据库搜索算法实现的,例如 BLAST(基本局部比对搜索工具),该算法执行目标序列与蛋白质序列数据库的序列比对。
PDB(蛋白质数据库)就是这样一种数据库。数据库中与我们的目标最匹配的蛋白质序列被认为是进化上最接近的,其结构将用作目标结构模型的模板。数据库搜索工具还给出了一个比对,即目标的哪些区域匹配模板的哪些区域的信息。
(二) 对齐校正
在数据库搜索期间获得的目标和模板之间的初始比对在比对的某些困难区域中可能不是最佳的。例如,初始比对可能违反某些氨基酸替换规则,例如用蛋白质核心中的疏水残基替换亲水残基。
鉴于我们已经找到了一个初始模板,我们现在可以使用更严格的对齐算法来找到更好的对齐方式。例如,我们可以在这一步使用多个序列比对算法。多序列比对可用于识别高度不同的区域,从而更好地检测插入和删除的适当位置。
(三) 骨干生成
优化目标模板比对后,生成目标的蛋白质骨架结构(N-Ca-Cb)。
这是通过基于对齐简单地将模板主干的坐标复制到目标来实现的。也就是说,目标蛋白质中原子的坐标与模板蛋白质中相应原子的坐标相同,如上一步的比对所述的那样。
这个过程高度依赖于模板结构的准确性,我们初始数据库中的任何错误都会导致我们预测的错误。
(四) 循环建模
主干步骤不处理对齐中存在的两种类型的不匹配,即插入和删除。将这些不匹配合并到主干中是同源建模中最困难的部分。
蛋白质的二级结构由螺旋、链和环(helices, strands and loops)组成。由于插入和删除所暗示的构象变化不能发生在螺旋和链中,它们必须发生在环中。
建模循环有两种主要方法:基于已知和基于能量。前一种方法在已知结构的数据库中搜索与目标具有相似序列和端点的环的构象。后者通过使用力场函数和分子动力学预测具有最低结构能量的环结构,以从头开始的方式对环构象进行建模。这些方法为最多 5-8 个残基的短环提供了相当准确的结果。
(五) 侧链建模
侧链建模涉及预测连接到主链的每个 R 基团的Ca-Cb扭转角(torsion angle)的值。
结构中侧链的构象,也称为旋转异构体,取决于该扭转角的值。侧链通常使用旋转异构体库的方式建模,旋转异构体库包含各种化学邻域下所有 20 个 R 基团的优选构象。
(六) 模型优化
既然蛋白质结构的所有方面都针对目标进行了建模,现在是对结构进行细微的改变以降低整体能量了。这是以迭代方式实现的。
在每次迭代中,主链构象和旋转异构体构象交替变化以降低预测结构的总能量。
模型优化也可以通过运行分子动力学模拟来执行,该模拟从当前预测的结构开始,并根据模拟对结构进行小的更改,即模拟在力作用下蛋白质的每个原子会发生什么在飞秒 (10 -15) 时间尺度上围绕它。
(七) 模型验证
最后一步是检查预测的结构是否有错误。由于目标和模板之间的低对齐或由于模板结构中的错误,在预测的蛋白质结构中引入了错误。
对预测的结构进行检查,看看是否所有的键长、键角和扭转角都落在从实验确定的蛋白质结构中发现的特征范围内。还执行能量检查,以查看不同类型的基于结构的能量(如范德华力和静电力)是否处于预期水平。
使用 SWISS-MODEL 进行同源建模
我们将通过使用SWISS-MODEL工具预测鸟氨酸氨甲酰转移酶 Ornithine carbamoyltransferase (UniProtKB accession: P96134) 中存在的蛋白质鸟氨酸氨基甲酰基转移酶的结构来详细研究同源建模程序。
目标模板识别
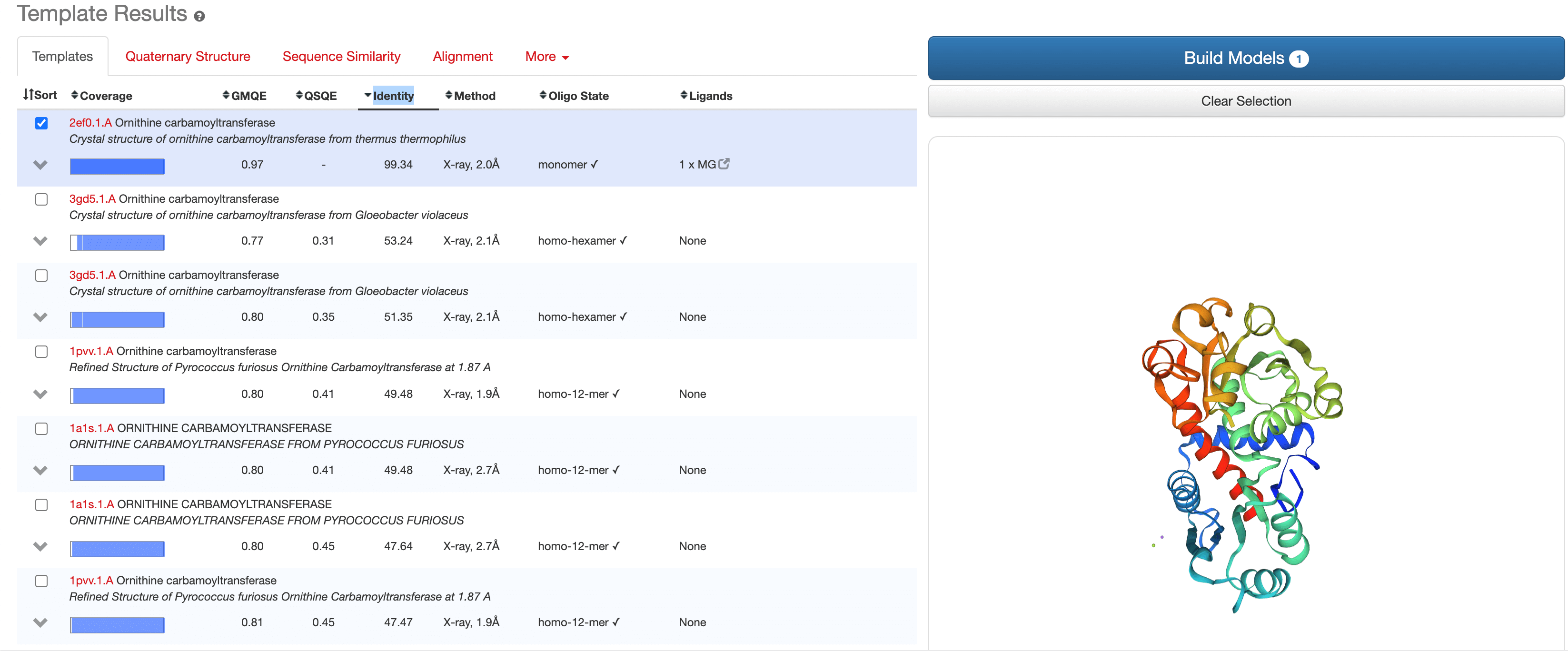
第一步是在已知蛋白质结构的序列数据库中搜索目标序列。将登录号粘贴到窗口中,然后点击 “Search For Templates” 按钮
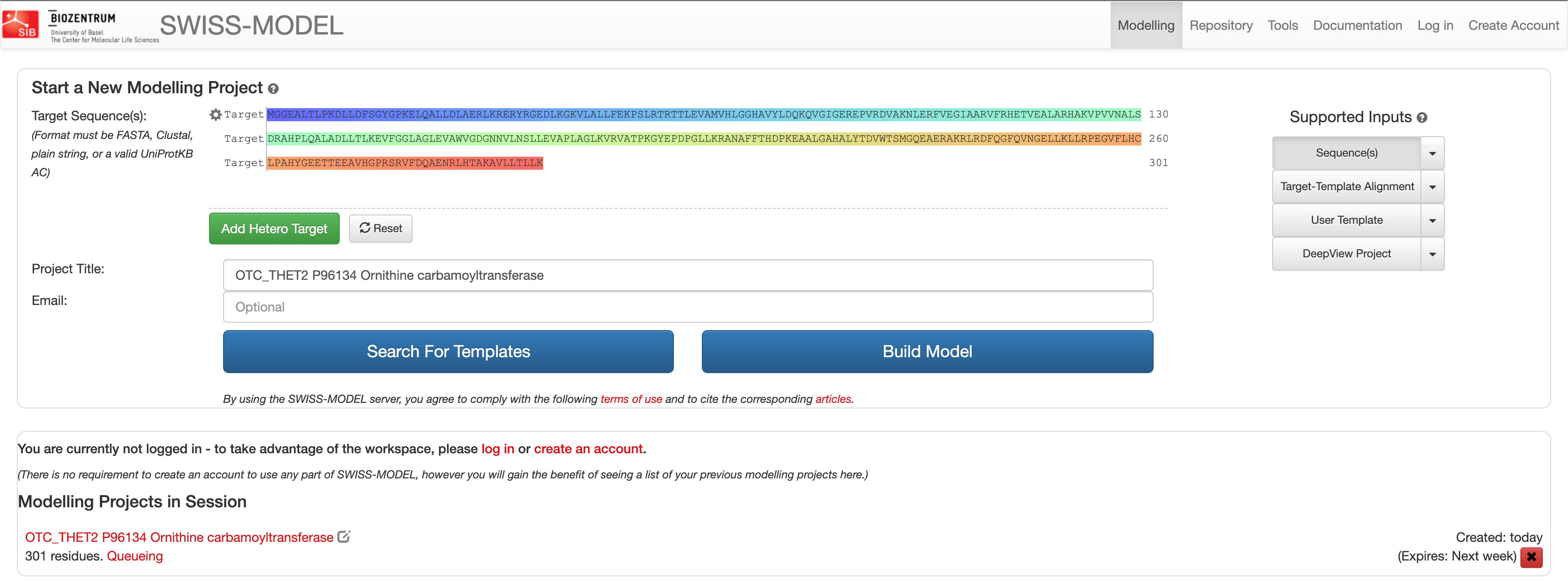
搜索结果显示了不同的蛋白质结构模板,可用于预测目标序列的蛋白质结构。这些模板根据其序列与目标蛋白质序列的对齐程度进行排序。
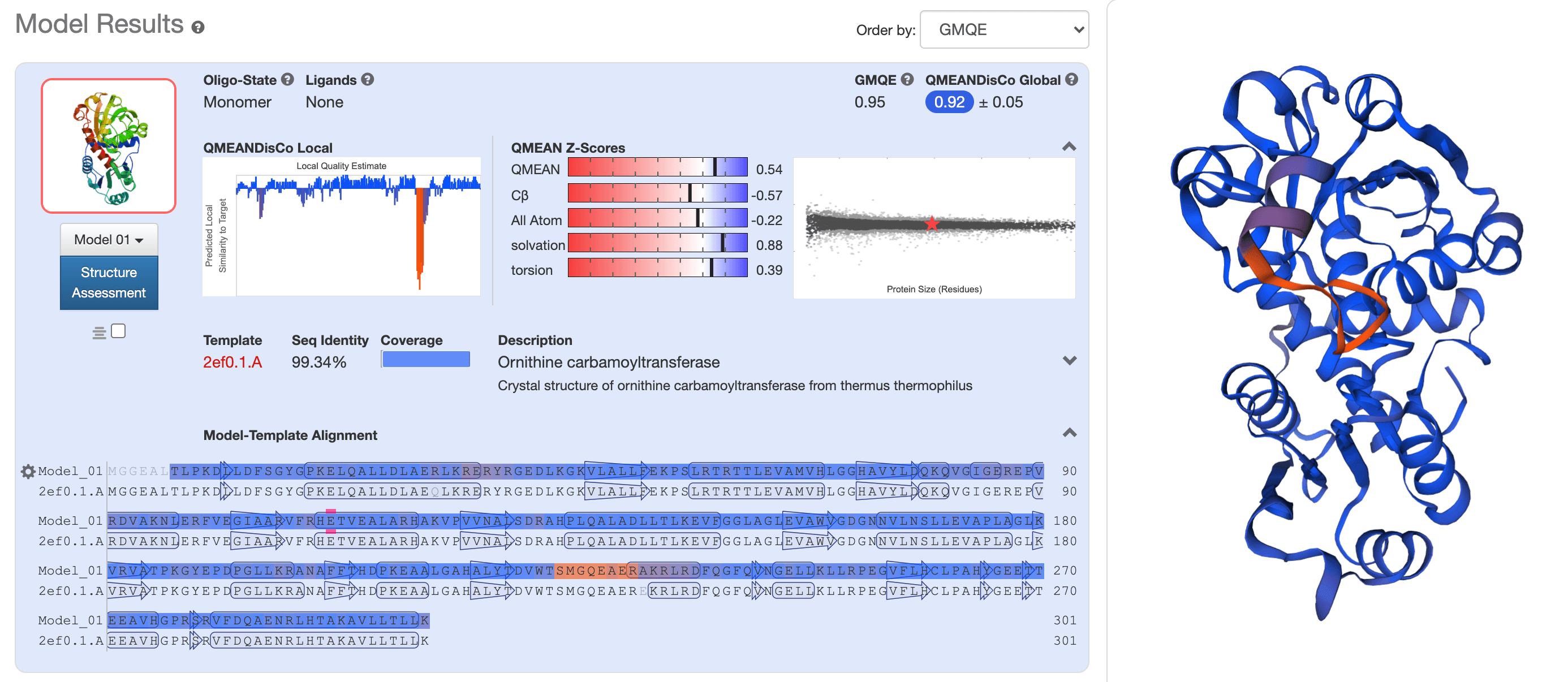
第一个选择的结构模板是最佳匹配(99% identity)。第二个模板与目标序列匹配,具有 53% identity。在右侧的窗口中可以看到两个顶部匹配模板的叠加蛋白质结构。
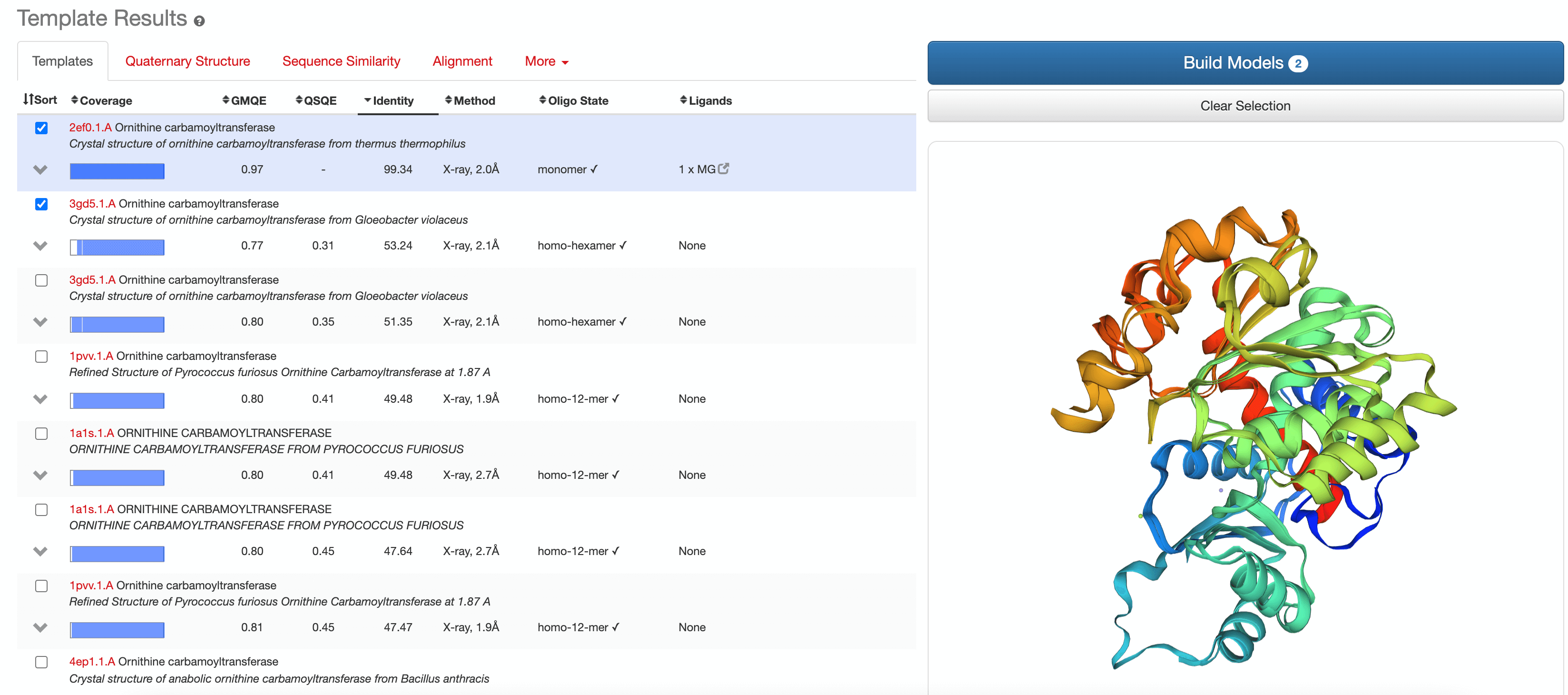
我们将使用前两个结果为目标序列构建或预测两个结构,然后选择最佳预测结构。
建模结果如下所示。根据模型的质量对这两个预测结构进行排名。
接下来,我们将评估两个预测结构的质量,看看哪个是最好的。
QMEAN 是用于评估模型质量的主要措施之一。QMEAN 是一种基于蛋白质结构不同几何特性的复合评分函数,提供全局(即整个结构)和局部(即每个残基)绝对质量估计。
QMEAN 由四个单独的术语组成。还列出了全局 QMEAN 质量分数的四个单独术语。条形图中的白色区域(数值接近于零)表明该特性与在实验结构中观察到的相似。正值表示模型平均得分高于实验结构,负数表示模型平均得分低于实验结构。
对于第一个模型(使用 2ef0.1.A 作为模板构建),QMEAN 项主要落在白色区域内。
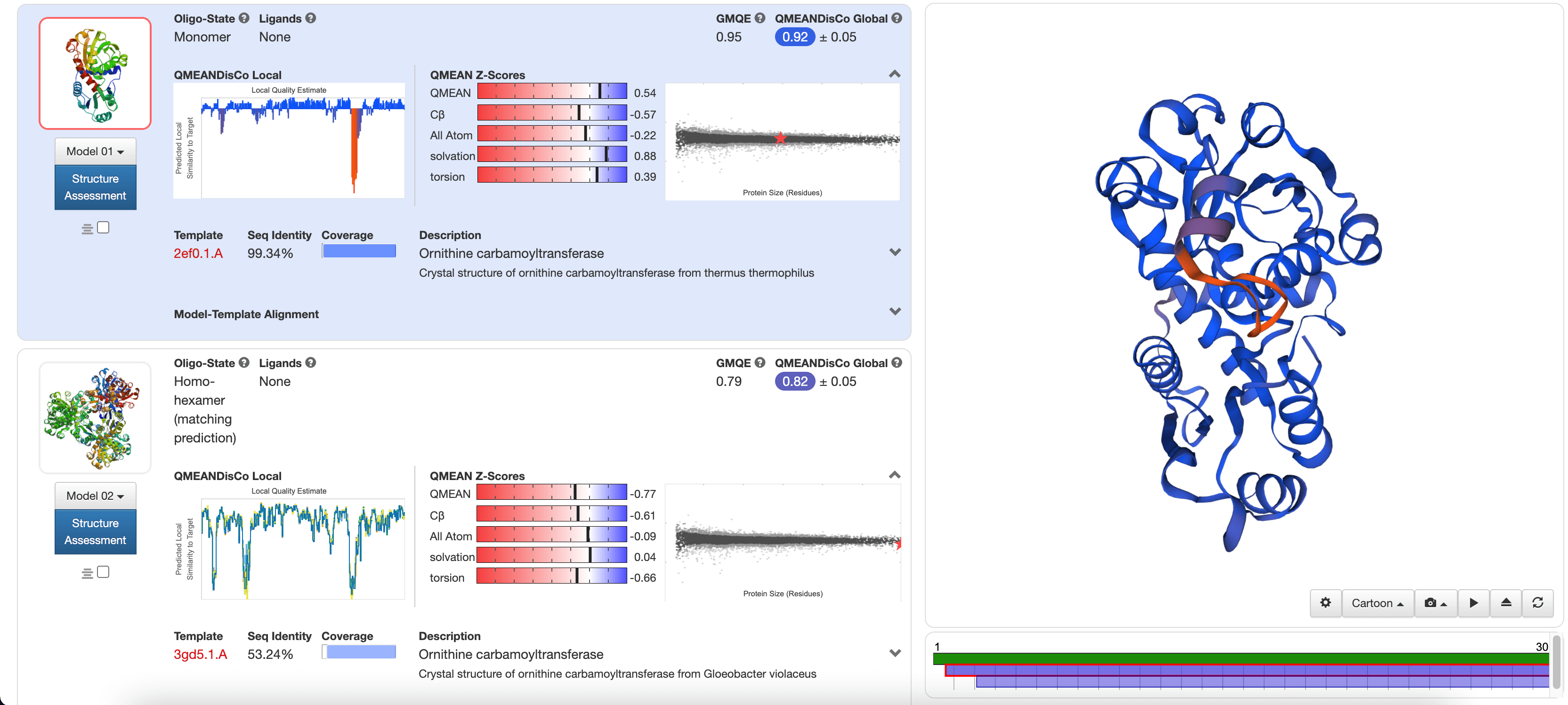
但是,对于第二个模型(使用 3gd5.1.A 作为模板构建),大多数 QMEAN 项与最优模型显著不同
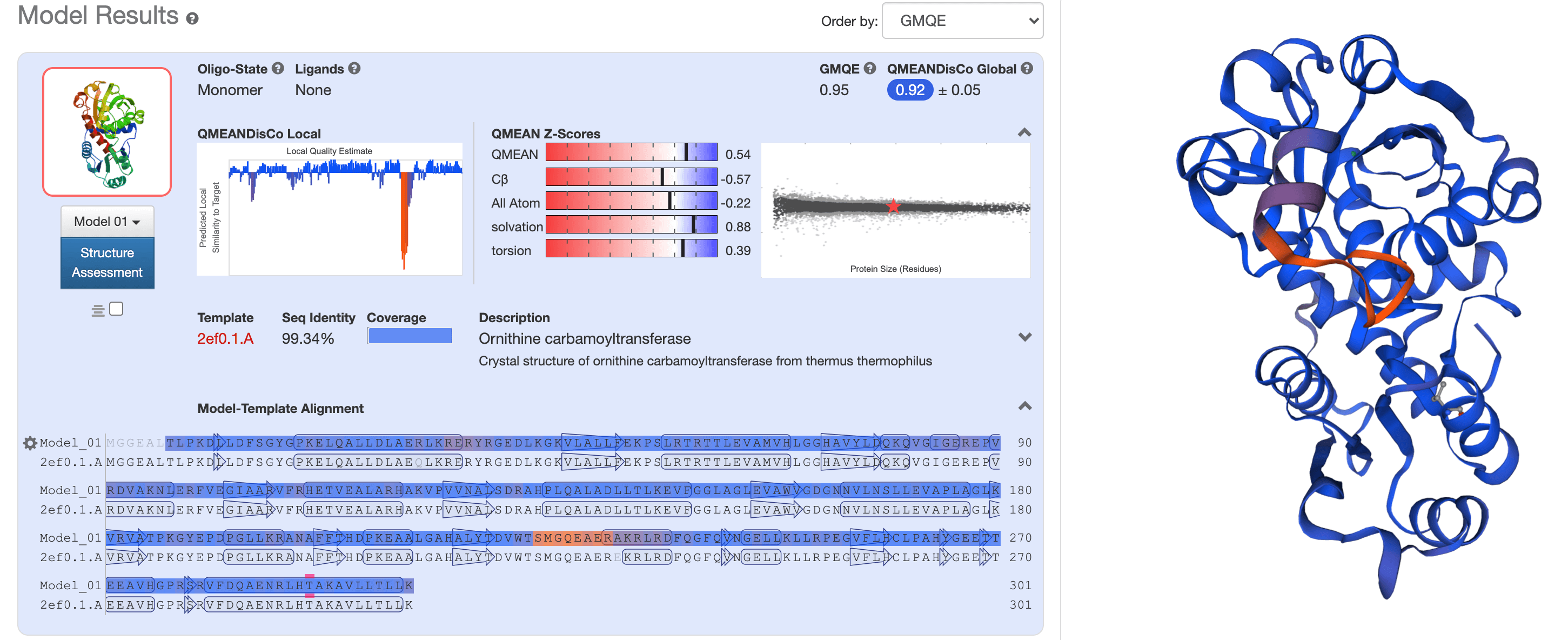
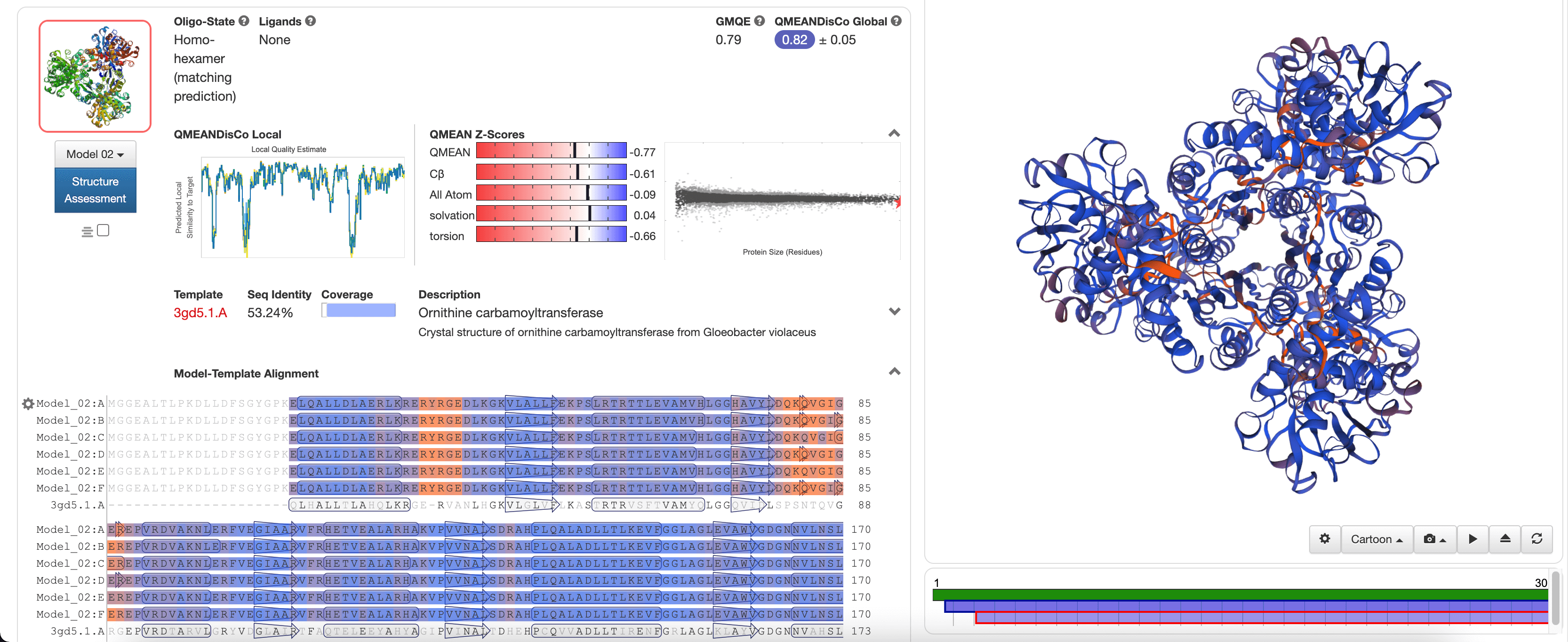
因此,模板2ef0.1.A预测的结构是最优模型,可以作为我们目标序列的预测结构。
参考
-
Swiss-MODEL https://swissmodel.expasy.org/interactive
-
MODELLER https://salilab.org/modeller/
-
“Homology Modeling” by Elmar Krieger, Sander B. Nabuurs, and Gert Vriend
 微信
微信 支付宝
支付宝





