Gviz包介绍
Gviz软件包简介
Gviz软件包旨在提供一个结构化的可视化框架,以沿着基因组坐标绘制任何类型的数据。它还允许整合来自UCSC 或ENSEMBL 等来源的公开基因组注释数据。
与大多数基因组浏览器一样,单独类型的基因组特征或数据由单独的Track 表示。
默认情况下,Gviz 检查所有提供的染色体名称在 UCSC 上的有效性(染色体必须以 chr 字符串开头)。options(ucscChromosomeNames=FALSE) 来关此功能
在以下示例中,将利用小鼠 mm9 基因组上的 UCSC 基因组和 7 号染色体 (chr7)
绘制 AnnotationTrack 图
请注意,AnnotationTrack
例如,注释功能的开始和结束坐标可以作为单个参数
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 library(Gviz) library(GenomicRanges) data(cpgIslands) cpgIslands
1 2 3 atrack <- AnnotationTrack(cpgIslands, name = "CpG" ) p<-plotTracks(atrack)
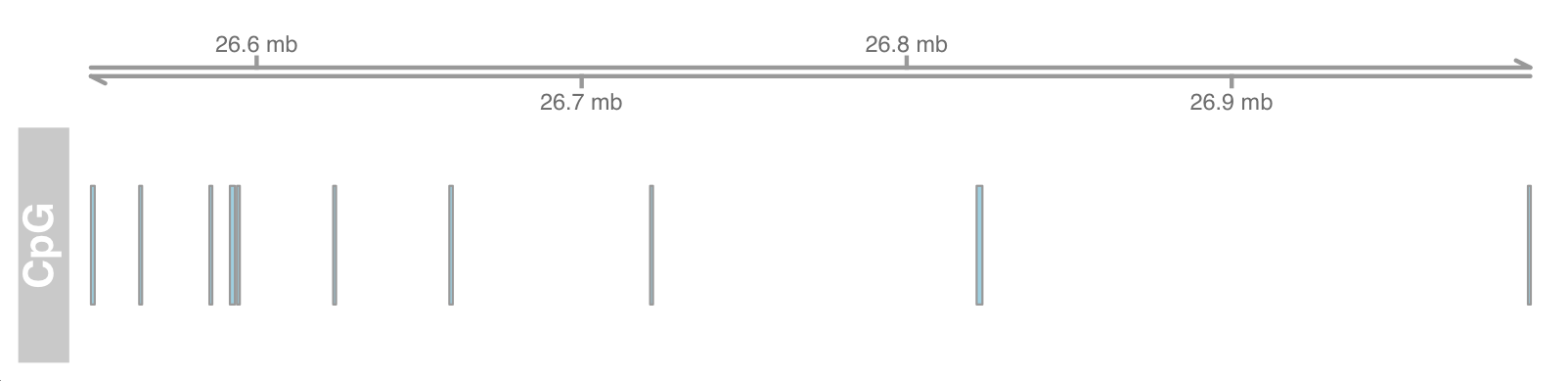
添加基因组坐标
1 2 3 gtrack <- GenomeAxisTrack() plotTracks(list (gtrack, atrack))
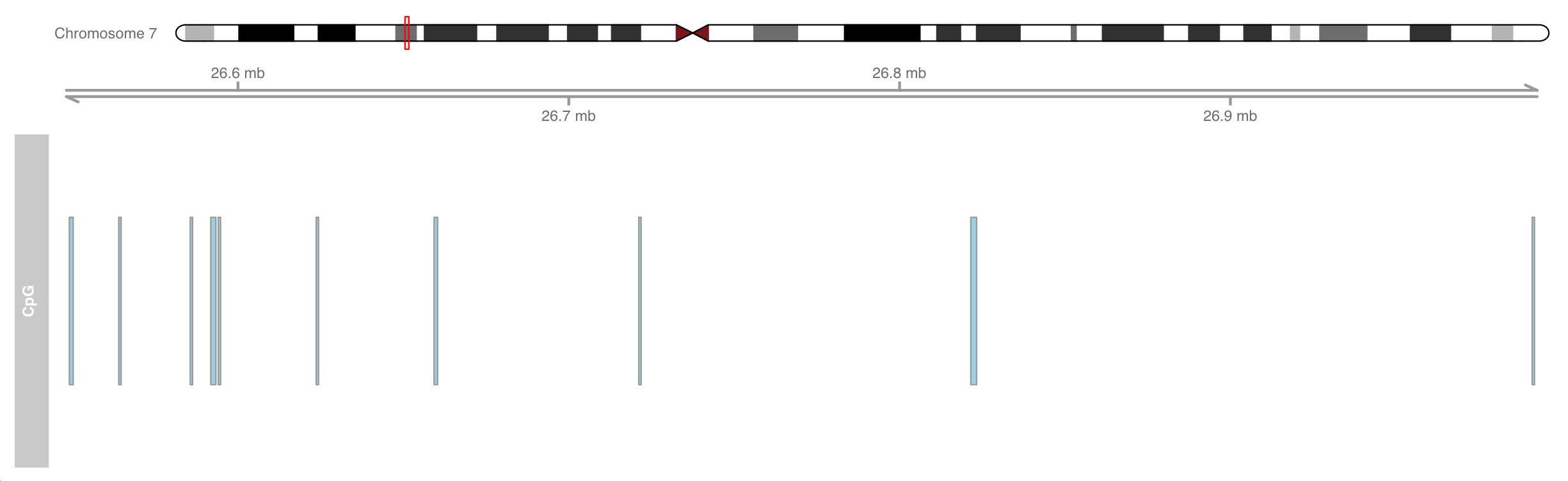
添加染色体表意文字
要添加染色体表意文字 (ideogram ),我们必须指明有效的 UCSC 基因组(例如:“hg19”)以及染色体名称(例如:“chr7”)
由于该功能从 UCSC 获取数据,因此需要 Internet 连接,这可能需要很长时间。
1 2 3 4 5 6 7 gen<-genome(cpgIslands) chr <- as.character (unique(seqnames(cpgIslands))) itrack <- IdeogramTrack(genome = gen, chromosome = chr) plotTracks(list (itrack, gtrack, atrack))
表意文字轨迹( ideogram track ) 是所有Gviz轨迹对象中的一个例外,因为它们不像所有其他轨迹那样真正显示在同一坐标系上。红色方框表示 (或者,在本例中,如果宽度太小而无法容纳方框,则用红线表示)。
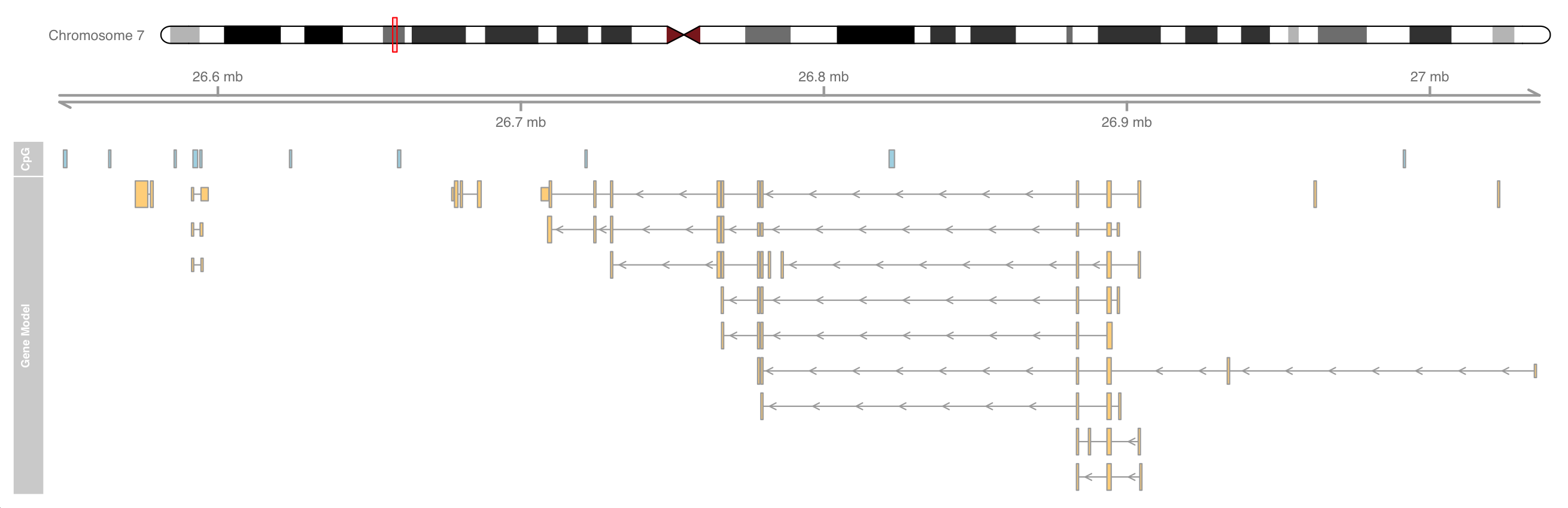
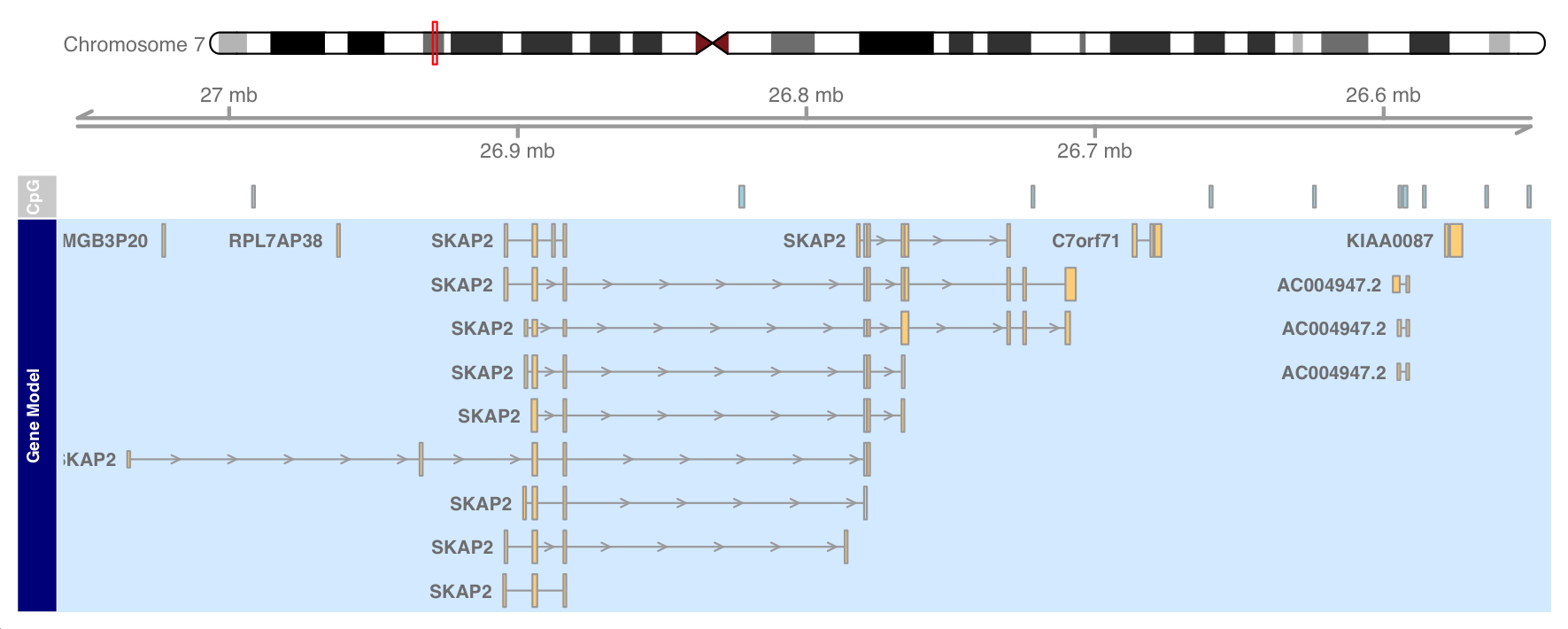
添加基因模型
我们可以利用现有本地来源的基因模型信息。或者,可以从可用的在线资源之一(如 UCSC 或 ENSEBML )下载此类数据,并且有构造函数来处理这些任务。
对于这个例子,我们将从存储的 data.frame 加载基因模型数据。
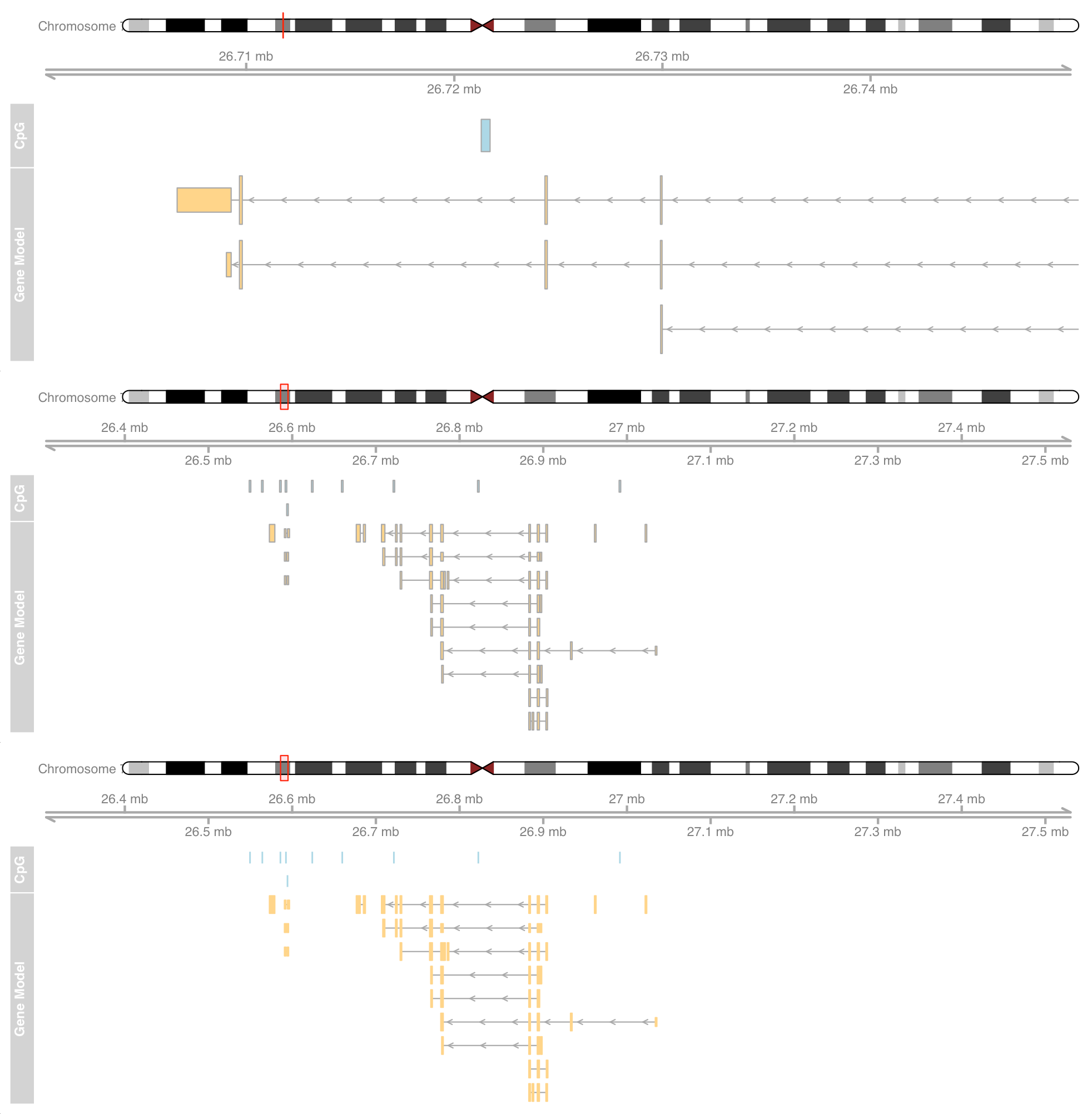
1 2 3 4 5 6 7 8 data(geneModels) head(geneModels) grtrack <- GeneRegionTrack(geneModels, genome = gen, chromosome = chr, name = "Gene Model" ) plotTracks(list (itrack, gtrack, atrack, grtrack))
放大绘图
我们经常想放大或缩小特定的绘图区域 以查看更多细节或获得更广泛的概览。
1 2 3 4 5 6 7 8 9 10 11 12 13 plotTracks(list (itrack, gtrack, atrack, grtrack), from = 26700000 , to = 26750000 ) plotTracks(list (itrack, gtrack, atrack, grtrack), extend.left = 0.5 , extend.right = 500000 ) plotTracks(list (itrack, gtrack, atrack, grtrack), extend.left = 0.5 , extend.right = 500000 , col = NULL )
值为 0.5 时将放大到当前显示范围的一半
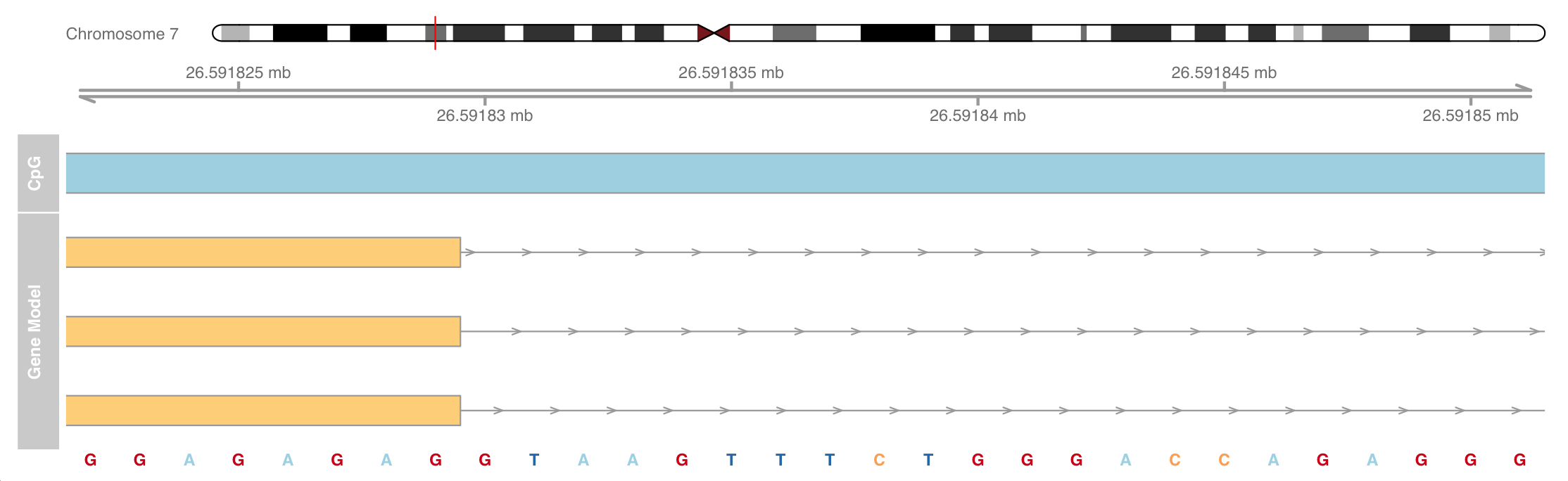
添加序列track并放大查看序列
必要的序列信息来自 BSgenome 包
1 2 3 4 5 6 7 8 if (!requireNamespace("BiocManager" , quietly = TRUE )) install.packages("BiocManager" ) BiocManager::install("BSgenome.Hsapiens.UCSC.hg19" ) library(BSgenome.Hsapiens.UCSC.hg19) strack <- SequenceTrack(Hsapiens, chromosome = chr) plotTracks(list (itrack, gtrack, atrack, grtrack, strack), from = 26591822 , to = 26591852 , cex = 0.8 )
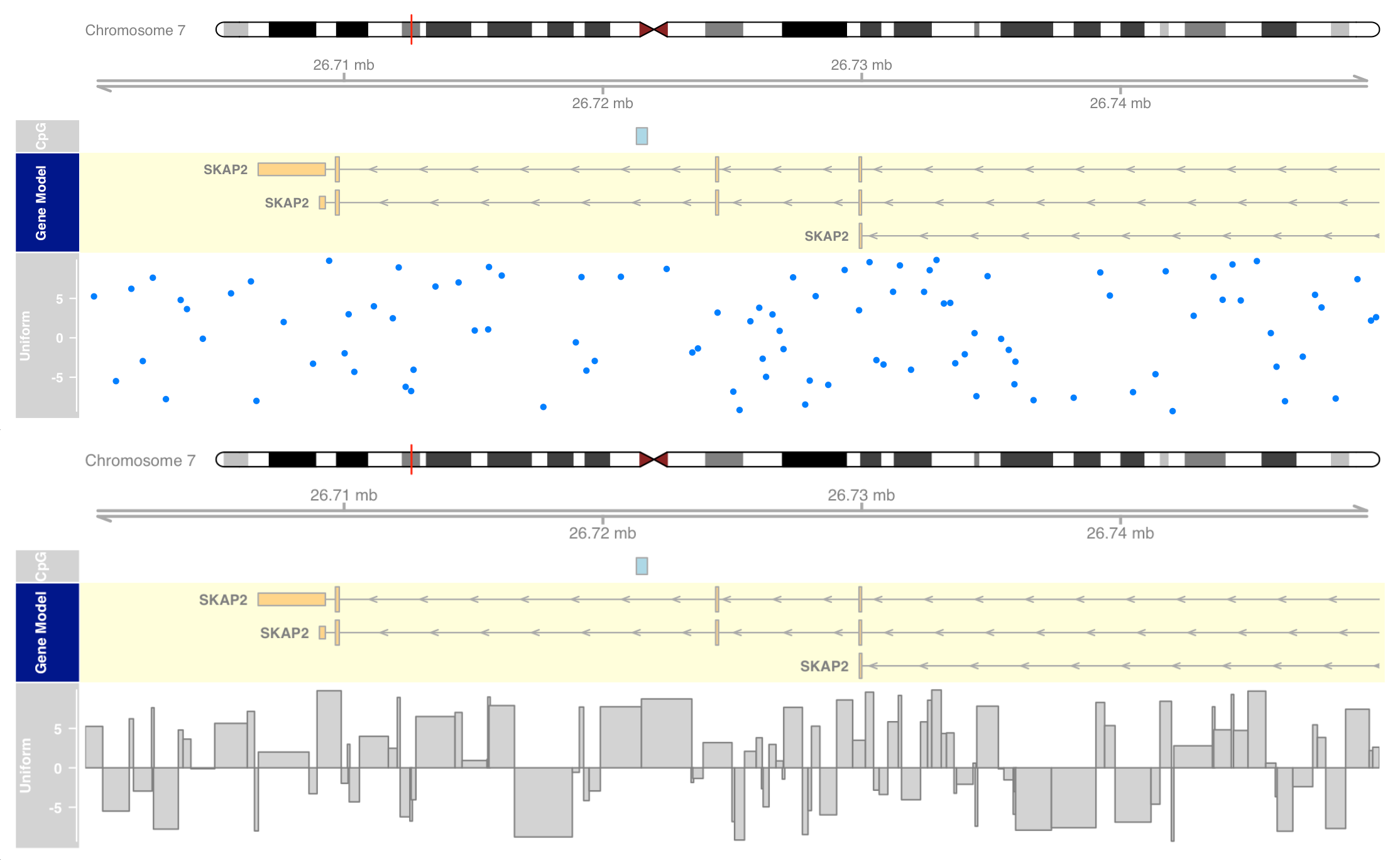
添加data track
DataTrack 对象本质上是运行长度编码 (Rle) 数字向量或矩阵,我们可以使用它们将各种数字数据添加到我们的基因组坐标图中。
这些 track 的不同可视化选项可用,包括点图、直方图和盒须图表示。
1 2 3 4 5 6 7 set.seed(255 ) lim <- c (26700000 , 26750000 ) coords <- sort(c (lim[1 ], sample(seq(from = lim[1 ], to = lim[2 ]), 99 ), lim[2 ])) dat <- runif(100 , min = -10 , max = 10 ) head(dat)
1 2 3 4 5 6 7 8 9 10 11 12 dtrack <- DataTrack(data = dat, start = coords[-length (coords)], end = coords[-1 ], chromosome = chr, genome = gen, name = "Uniform" ) plotTracks(list (itrack, gtrack, atrack, grtrack, dtrack), from = lim[1 ], to = lim[2 ]) plotTracks(list (itrack, gtrack, atrack, grtrack,dtrack), from = lim[1 ], to = lim[2 ], type = "histogram" )
这种可视化在显示例如沿染色体的 NGS reads 的覆盖范围时特别有用,或显示来自微阵列实验的映射探针的测量值。
绘图参数
设置参数
1 2 3 4 5 6 7 8 grtrack <- GeneRegionTrack(geneModels, genome = gen, chromosome = chr, name = "Gene Model" , transcriptAnnotation = "symbol" , background.panel = "#dbeeff" , background.title = "darkblue" ) plotTracks(list (itrack, gtrack, atrack, grtrack))
绘图方向
默认情况下,所有轨迹都将以 5’ -> 3’ 方向绘制。有时实际显示相对于相反链的数据会很有用。
1 2 plotTracks(list (itrack, gtrack, atrack, grtrack), reverseStrand = TRUE )
数据已绘制在反向链上这也反映在 GenomeAxis track 中。
Track classes
多个参数可用于更改不同轨道的外观。
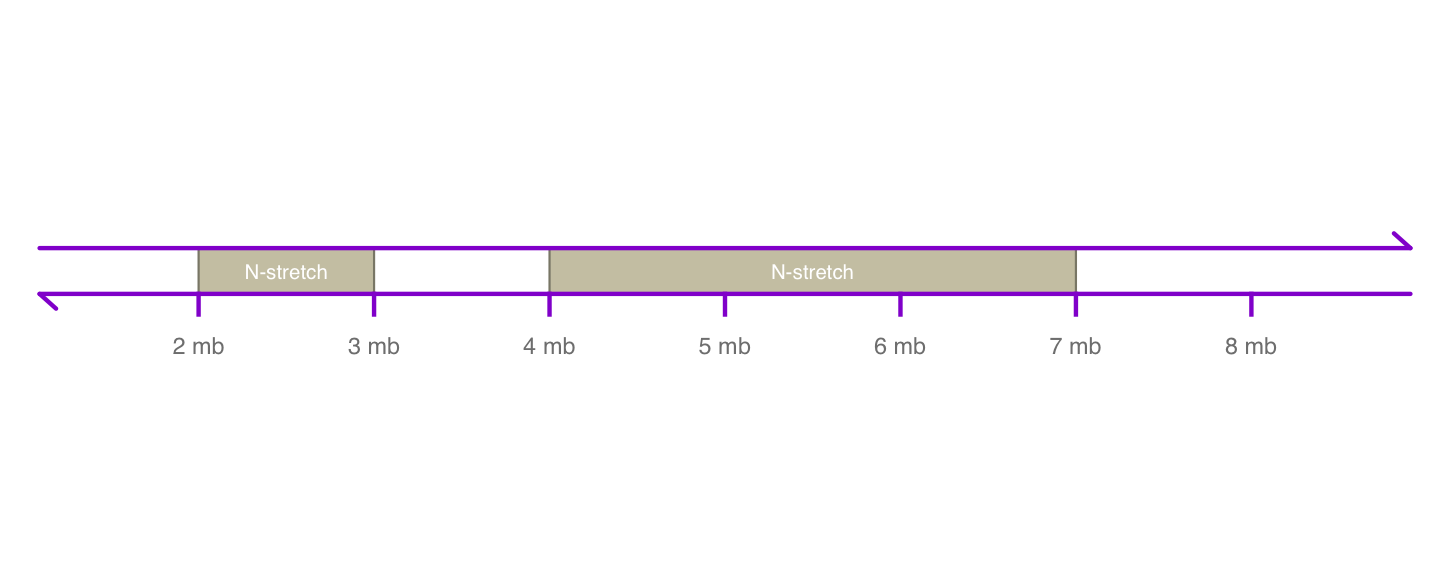
GenomeAxisTrack 的显示参数
将标签的位置设置为下方,显示 ID,更改颜色
1 2 3 axisTrack <- GenomeAxisTrack(range = IRanges(start = c (2000000 ,4000000 ), end = c (3000000 , 7000000 ), names = rep ("N-stretch" , 2 ))) plotTracks(axisTrack, from = 1000000 , to = 9000000 , labelPos = "below" ,showId=TRUE , col="#9400D3" )
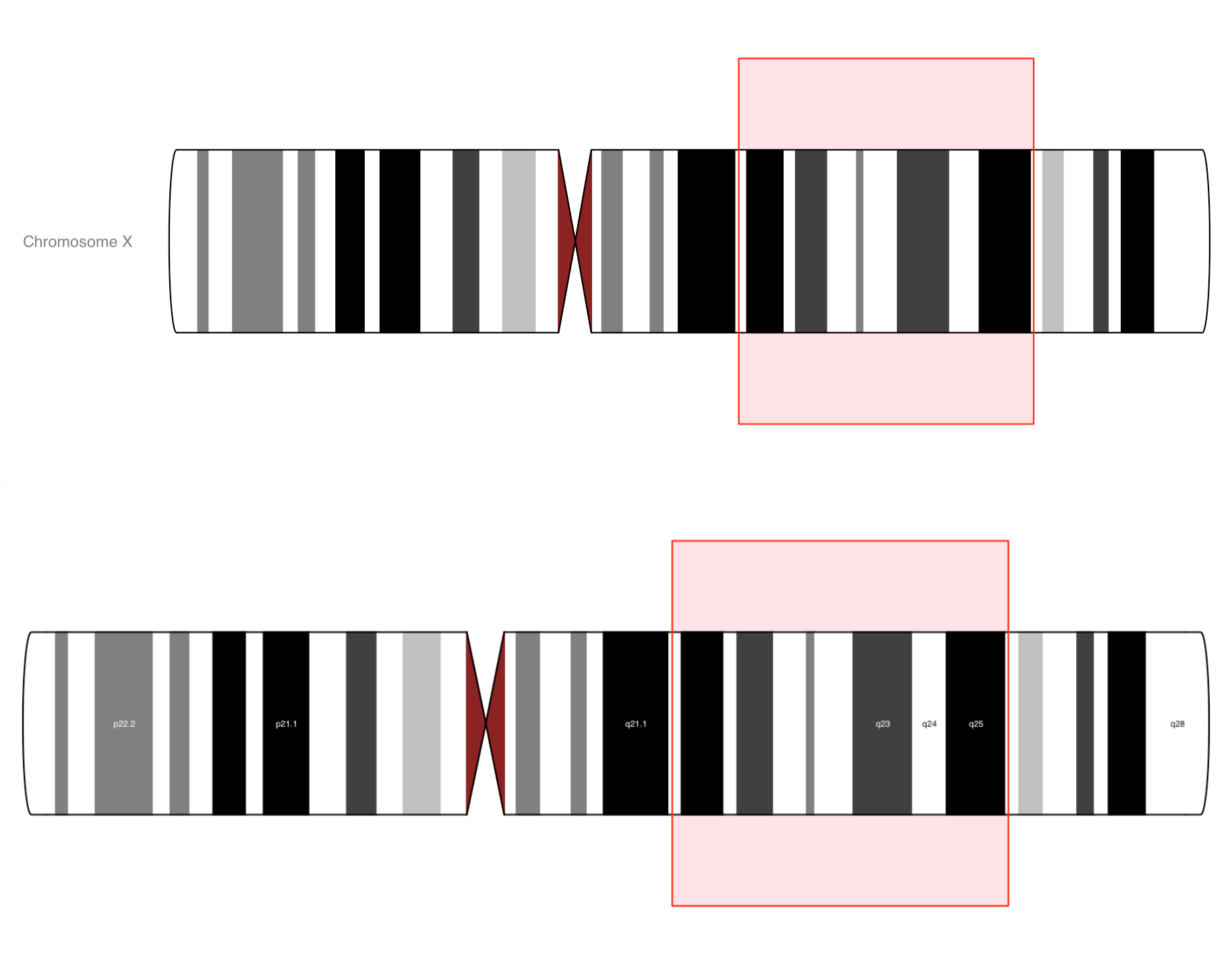
IdeogramTrack
1 2 3 4 5 6 7 ideoTrack <- IdeogramTrack(genome = "hg19" , chromosome = "chrX" ) plotTracks(ideoTrack, from = 85000000 , to = 129000000 ) plotTracks(ideoTrack, from = 85000000 , to = 129000000 , showId = FALSE , showBandId = TRUE , cex.bands = 0.4 )
DataTrack
本质上,它们构成了与特定基因组坐标范围相关联的运行长度编码的数字向量或矩阵。
单个数据集中可以有多个样本,绘图方法提供了合并样本组信息的工具。
因此,创建 DataTrack 对象的起点将始终是一组范围,以 IRanges 或 GRanges 对象的形式,或单独作为开始和结束坐标或宽度。第二个成分是长度与范围数相同的数值向量,或具有相同列数的数值矩阵。
接下来将从作为 Gviz 包的一部分的 GRanges 对象加载我们的示例数据
1 2 3 data(twoGroups) head(twoGroups)
1 2 3 dTrack <- DataTrack(twoGroups, name = "uniform" ) plotTracks(dTrack)
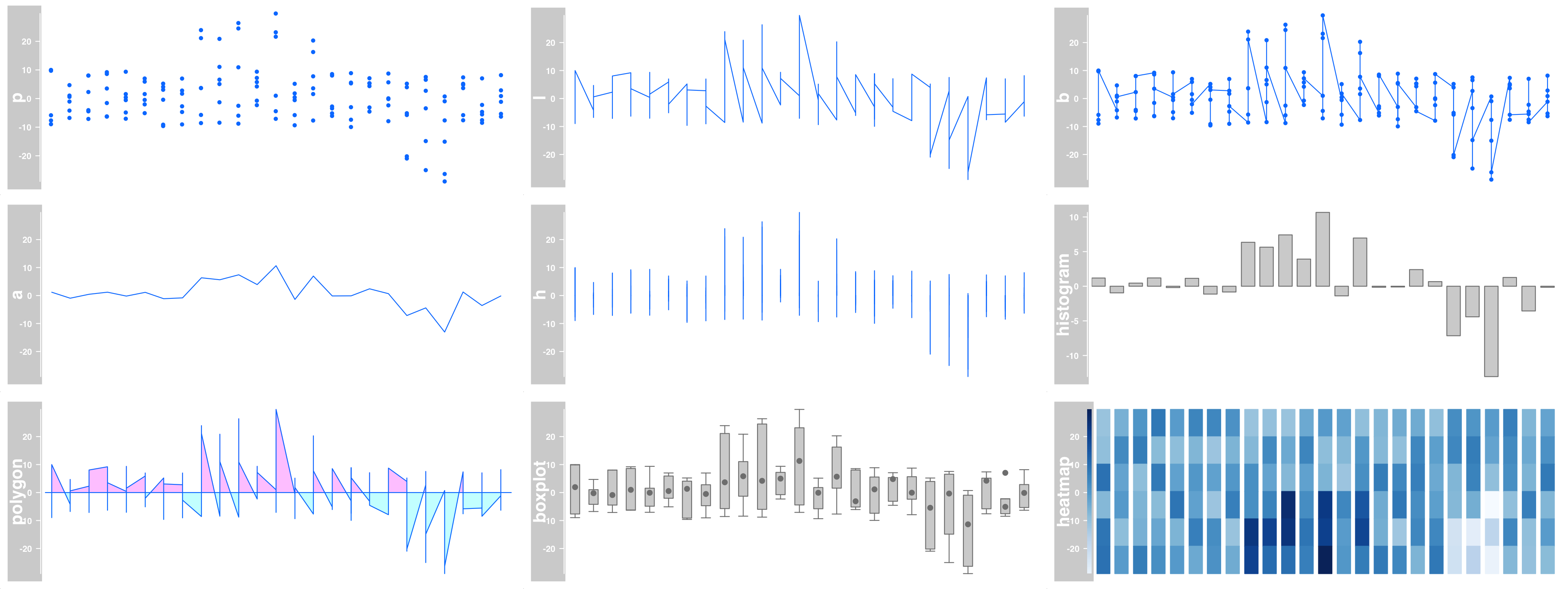
不同图片类型
可能的绘图类型是:
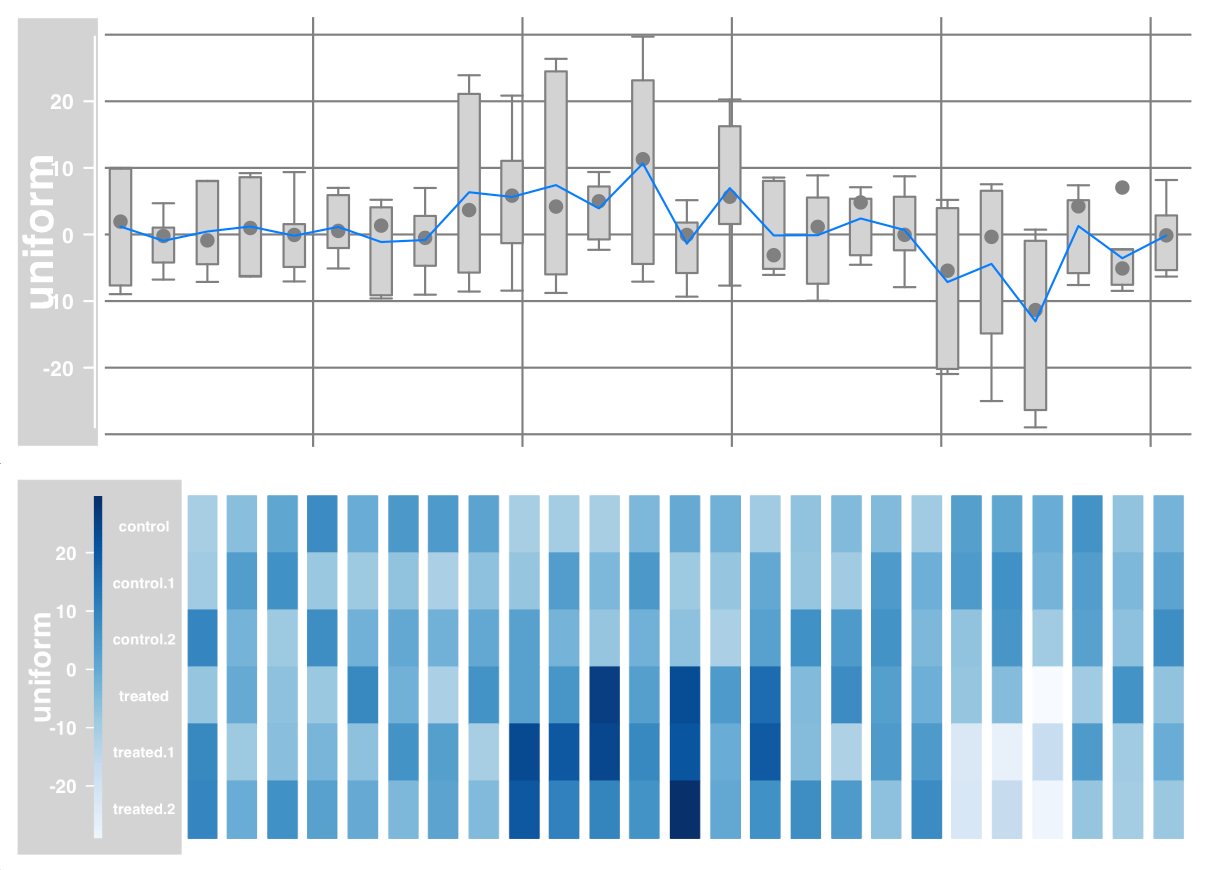
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 plotTracks(DataTrack(twoGroups, name = "p" ), type="p" ) plotTracks(DataTrack(twoGroups, name = "l" ), type="l" ) plotTracks(DataTrack(twoGroups, name = "b" ), type="b" ) plotTracks(DataTrack(twoGroups, name = "a" ), type="a" ) plotTracks(DataTrack(twoGroups, name = "h" ), type="h" ) plotTracks(DataTrack(twoGroups, name = "histogram" ), type="histogram" ) plotTracks(DataTrack(twoGroups, name = "polygon" ), type="polygon" ) plotTracks(DataTrack(twoGroups, name = "boxplot" ), type="boxplot" ) plotTracks(DataTrack(twoGroups, name = "heatmap" ), type="heatmap" )
DataTrack 图示例:
1 2 3 4 5 plotTracks(dTrack, type = c ("boxplot" , "a" , "g" )) plotTracks(dTrack, type = c ("heatmap" ), showSampleNames = TRUE , cex.sampleNames = 0.6 )
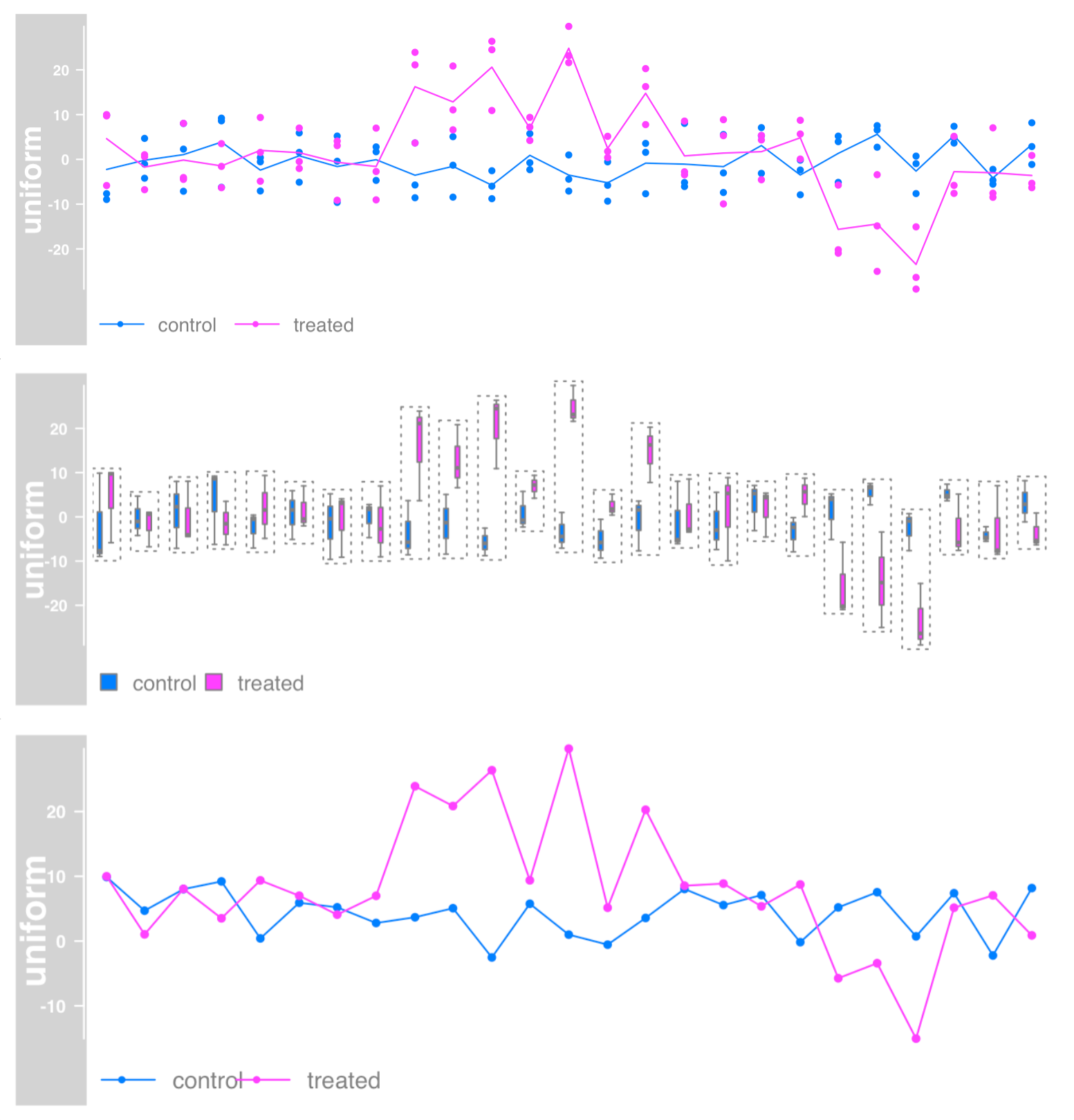
数据分组
可以使用因子变量将单个样本组合在一起。
1 2 3 4 5 6 7 8 9 10 plotTracks(dTrack, groups = rep (c ("control" , "treated" ), each = 3 ), type = c ("a" , "p" ), legend=TRUE ) plotTracks(dTrack, groups = rep (c ("control" , "treated" ), each = 3 ), type = "boxplot" ) plotTracks(dTrack, groups = rep (c ("control" , "treated" ),each = 3 ), type = c ("b" ), aggregateGroups = TRUE , aggregation = "max" )
从文件构建 DataTrack 对象
DataTrack wig、bigWig、bedGraph 和 bam 文件
1 2 3 4 bgFile <- system.file("extdata/test.bedGraph" , package = "Gviz" ) dTrack2 <- DataTrack(range = bgFile, genome = "hg19" , type = "l" , chromosome = "chr19" , name = "bedGraph" ) plotTracks(dTrack2)
请注意, Gviz 包使用 rtracklayer 包 中的功能进行大多数文件导入操作
Gviz 包中文件支持来自索引文件的流式传输。
我们将在此使用随包提供的小 bam 文件来举例说明此功能。
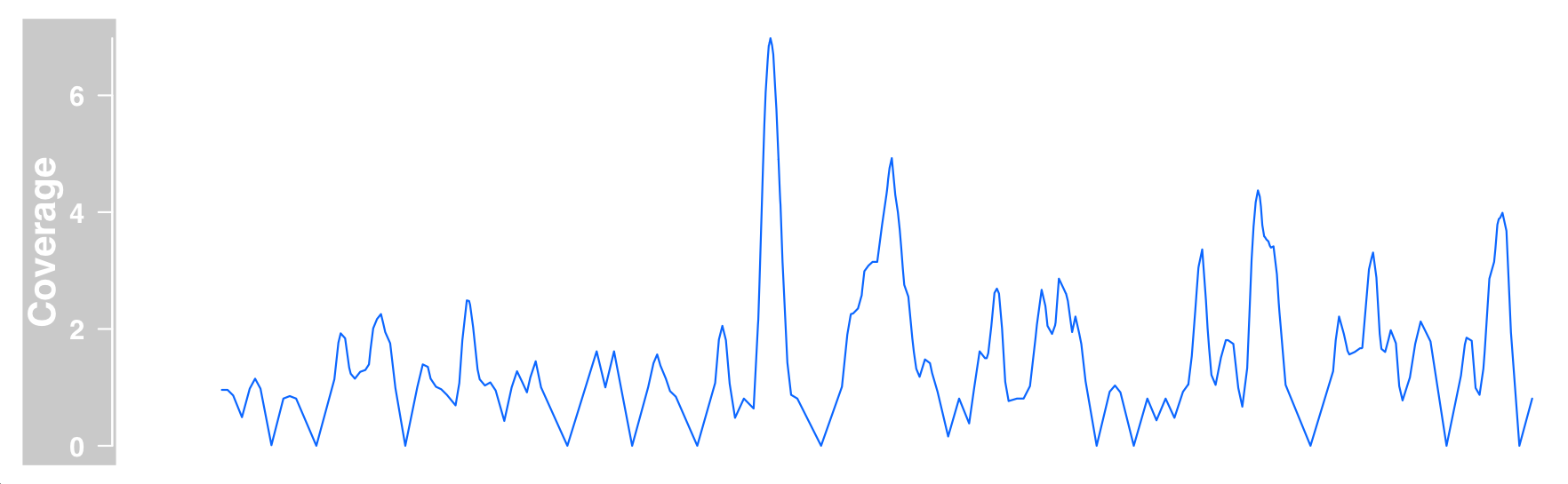
bam 文件包含序列与共同参考的比对。DataTrack 中此类数据的最自然表示是仅查看给定位置的对齐覆盖率,并将其编码在单个 elementMetadata 列中。
1 2 3 4 bamFile <- system.file("extdata/test.bam" , package = "Gviz" ) dTrack4 <- DataTrack(range = bamFile, genome = "hg19" , type = "l" , name = "Coverage" , window = -1 , chromosome = "chr1" ) plotTracks(dTrack4, from = 189990000 , to = 190000000 )
AnnotationTrack
本质上,它们由一个或多个基因组范围组成,如果需要,可以将这些范围分组为复合注释元素。
必要的构建块是范围坐标、染色体和基因组标识符。
信息可以以单独的函数参数的形式传递给函数,如 IRanges、GRanges 或 data.frame 对象。
1 2 3 4 5 aTrack <- AnnotationTrack(start = c (10 , 40 , 120 ), width = 15 , chromosome = "chrX" , strand = c ("+" ,"*" , "-" ), id = c ("Huey" , "Dewey" , "Louie" ), genome = "hg19" , name = "foo" ) plotTracks(aTrack)
从文件构建 AnnotationTrack 对象
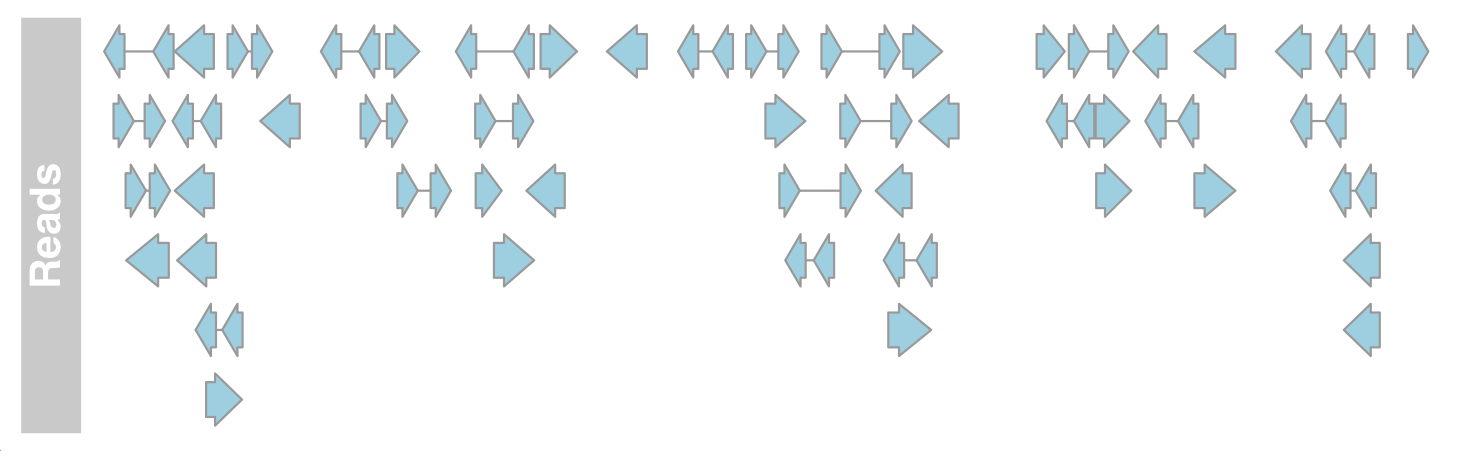
默认的导入函数从 bam 文件中读取所有序列比对的坐标。
1 2 3 4 aTrack2 <- AnnotationTrack(range = bamFile, genome = "hg19" , name = "Reads" , chromosome = "chr1" ) plotTracks(aTrack2, from = 189995000 , to = 190000000 )
我们现在可以同时绘制 bam 文件的 DataTrack 和 AnnotationTrack 表示,以证明基础数据确实相同。
1 2 plotTracks(list (dTrack4, aTrack2), from = 189990000 , to = 190000000 )
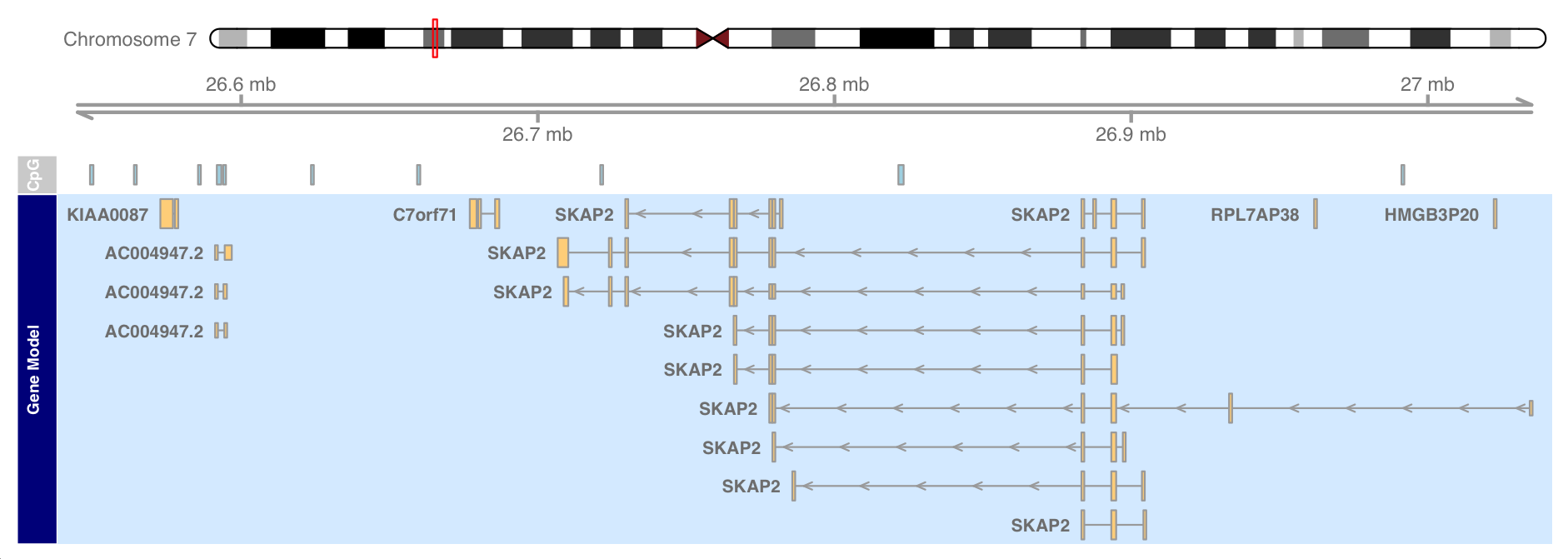
GeneRegionTrack
GeneRegionTrack 原则上与 AnnotationTrack 非常相似。
唯一的区别是它们更以基因/转录本为中心。我们需要传递track中每个注释特征的开始和结束位置(或宽度),并为每个项目提供外显子、转录本和基因标识符,这些标识符将用于创建转录本分组。
一个有点特殊的情况是直接从 TranscriptDb 之一构建 GeneRegionTrack ,下面将更详细地介绍该选项。
1 2 3 4 data(geneModels) grtrack <- GeneRegionTrack(geneModels, genome = gen, chromosome = chr, name = "foo" , transcriptAnnotation = "symbol" )
从 fTranscriptDbsx 构建 GeneRegionTrack 对象
GenomicFeatures 包 提供了一个框架,可以从在线资源下载基因模型信息并将其本地存储在 SQLite 数据库中。
从 TranscriptDb 构建 GeneRegionTracks 的一个好处是我们获得了关于转录本编码和非编码区域的额外信息,即 5’ 和 3’ UTR 以及 CDS 区域的坐标。
1 2 3 4 5 6 7 library(GenomicFeatures) samplefile <- system.file("extdata" , "UCSC_knownGene_sample.sqlite" , package = "GenomicFeatures" ) txdb <- loadDb(samplefile) txTr <- GeneRegionTrack(txdb, chromosome = "chr6" , start = 300000 , end = 350000 ) plotTracks(txTr)
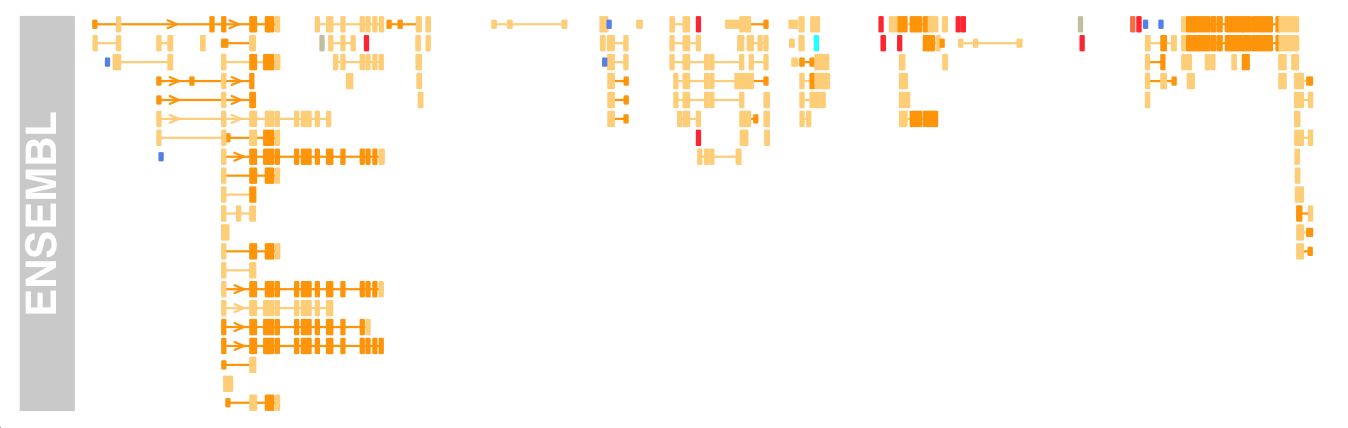
BiomartGeneRegionTrack BiomartGeneRegionTrack ENSEMBL Biomart 服务的直接接口。
我们只需输入基因组、染色体以及该染色体上的开始和结束位置,构造函数 BiomartGeneRegionTrack 将自动联系 ENSEMBL ,获取必要的信息并动态构建基因模型。
需要互联网连接才能工作,并且根据使用情况和网络流量,联系 Biomart 可能需要大量时间。
1 2 3 4 biomTrack <- BiomartGeneRegionTrack(genome = "hg19" , chromosome = chr, start = 20000000 , end = 21000000 , name = "ENSEMBL" ) plotTracks(biomTrack, col.line = NULL , col = NULL )
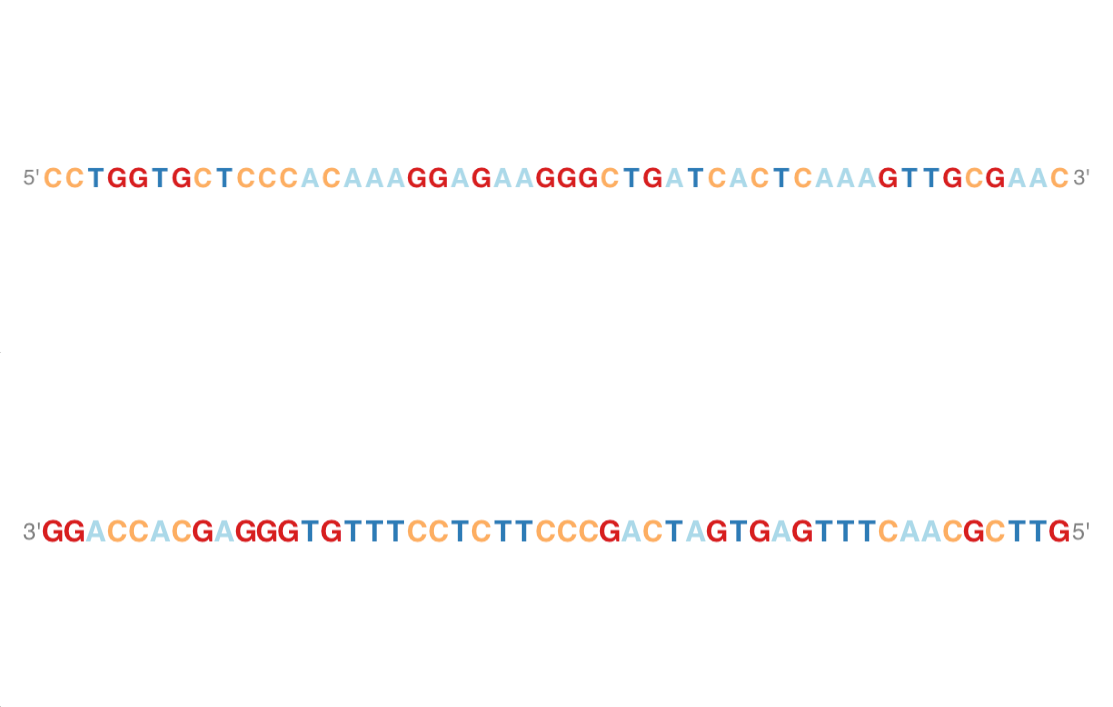
Sequence Track
1 2 3 4 5 6 7 8 library(BSgenome.Hsapiens.UCSC.hg19) sTrack <- SequenceTrack(Hsapiens) plotTracks(sTrack, chromosome = 1 , from = 20000 ,to = 20050 , add53=TRUE ) plotTracks(sTrack, chromosome = 1 , from = 20000 ,to = 20050 , add53=TRUE , complement = TRUE )
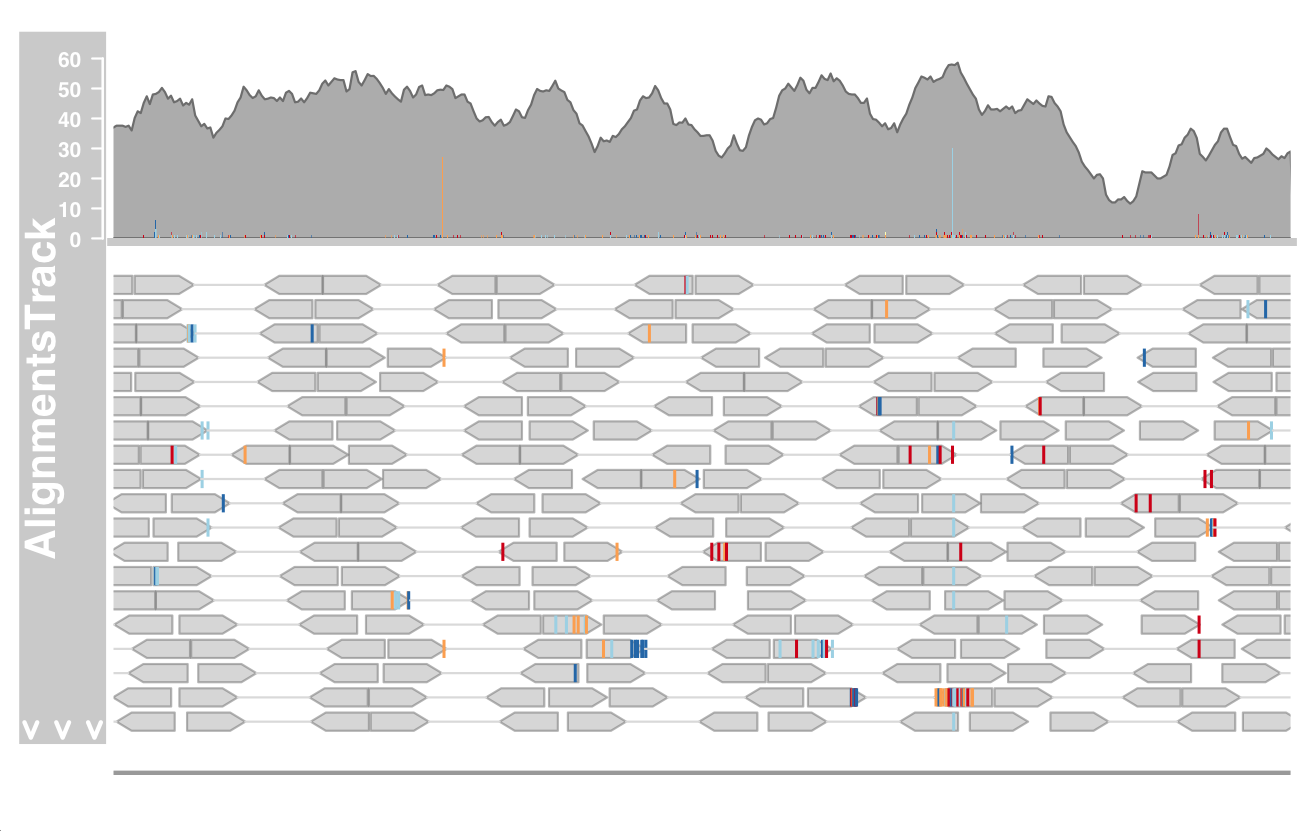
AlignmentsTrack
比对序列 (Aligned sequences ) 的图,通常来自下一代测序实验,可能非常有用,例如在目视检查称为 SNP 的有效性时。这些比对通常存储在 BAM 文件中。
RNA-seq
在本演示中,让我们使用一个小 BAM 文件,其中配对的 NGS reads 已映射到人类 hg19 基因组的提取物。
数据来自 RNA-Seq 实验,比对是使用 STAR 进行的,允许存在 gaps。
还从 Biomart 下载了该区域的一些基因注释数据。
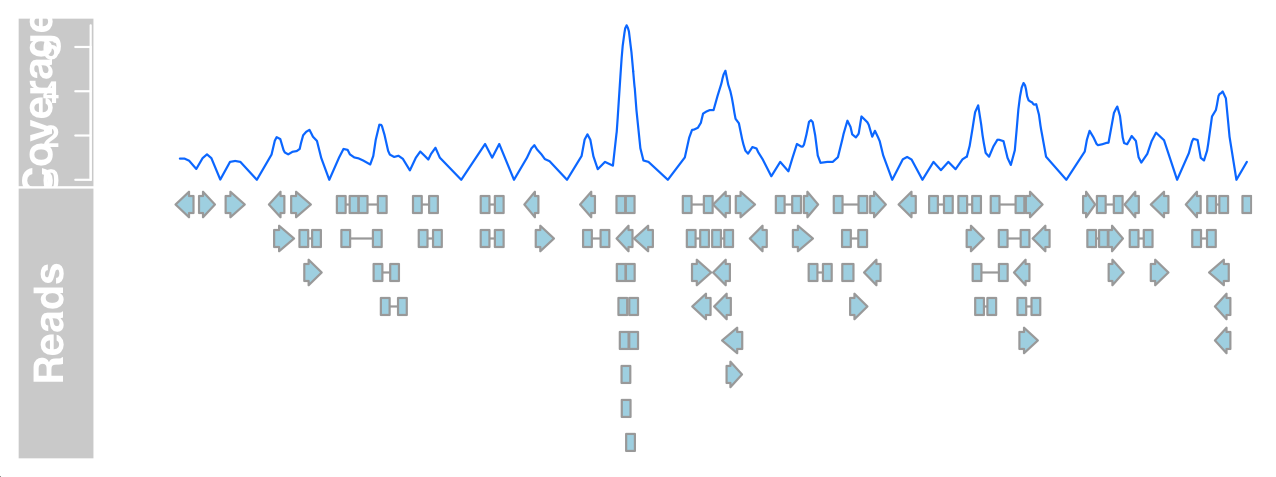
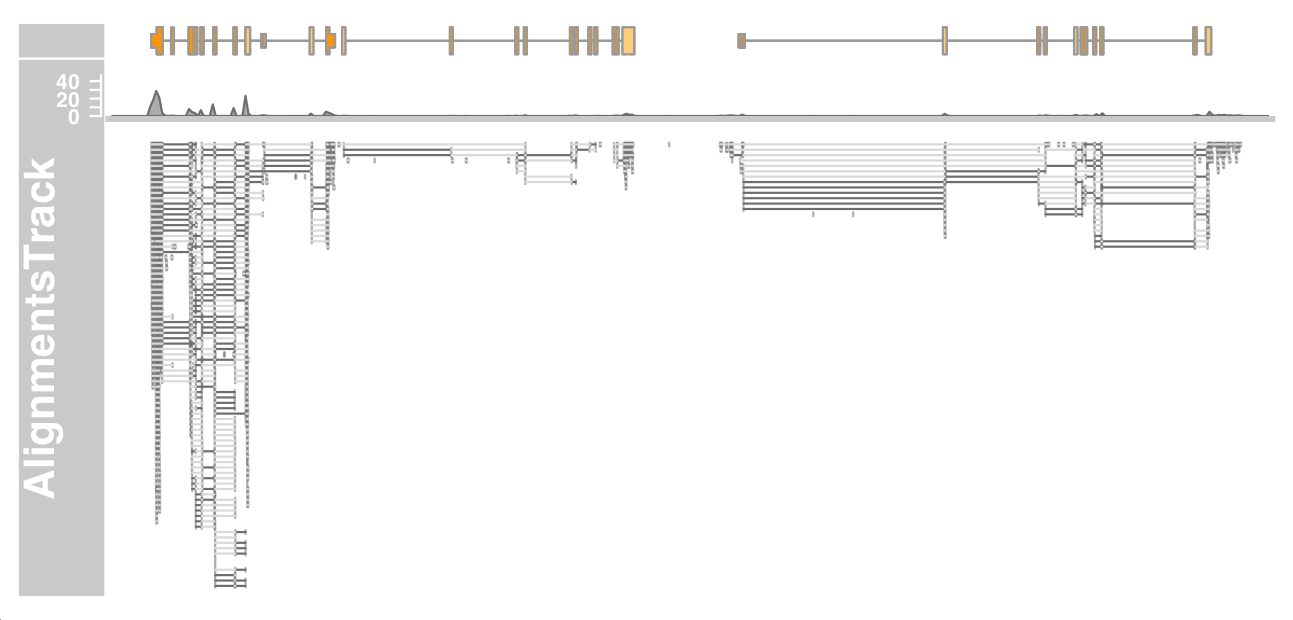
1 2 3 4 5 6 7 8 afrom=2960000 ato=3160000 alTrack <- AlignmentsTrack(system.file(package = "Gviz" , "extdata" , "gapped.bam" ), isPaired = TRUE ) bmt <- BiomartGeneRegionTrack(genome = "hg19" , chromosome = "chr12" , start = afrom, end = ato, filter = list (with_ox_refseq_mrna = TRUE ), stacking = "dense" ) plotTracks(c (bmt, alTrack), from = afrom, to = ato, chromosome = "chr12" )
上图展示了 track 的总体布局 在顶部 ,我们有一个以柱状图形式显示reads覆盖率信息的面板,在该面板下方 ,是单个 reads 的堆积视图。max.height、min.height或stackHeight显示参数来解决这个问题,这些参数都控制堆叠 reads 的高度或垂直间距。或者,我们可以通过设置 coverageHeight 或 minCoverageHeight 参数来减小覆盖区域的大小。
1 2 3 plotTracks(c (bmt, alTrack), from = afrom, to = ato, chromosome = "chr12" , min.height = 0 , coverageHeight = 0.08 , minCoverageHeight = 0 )
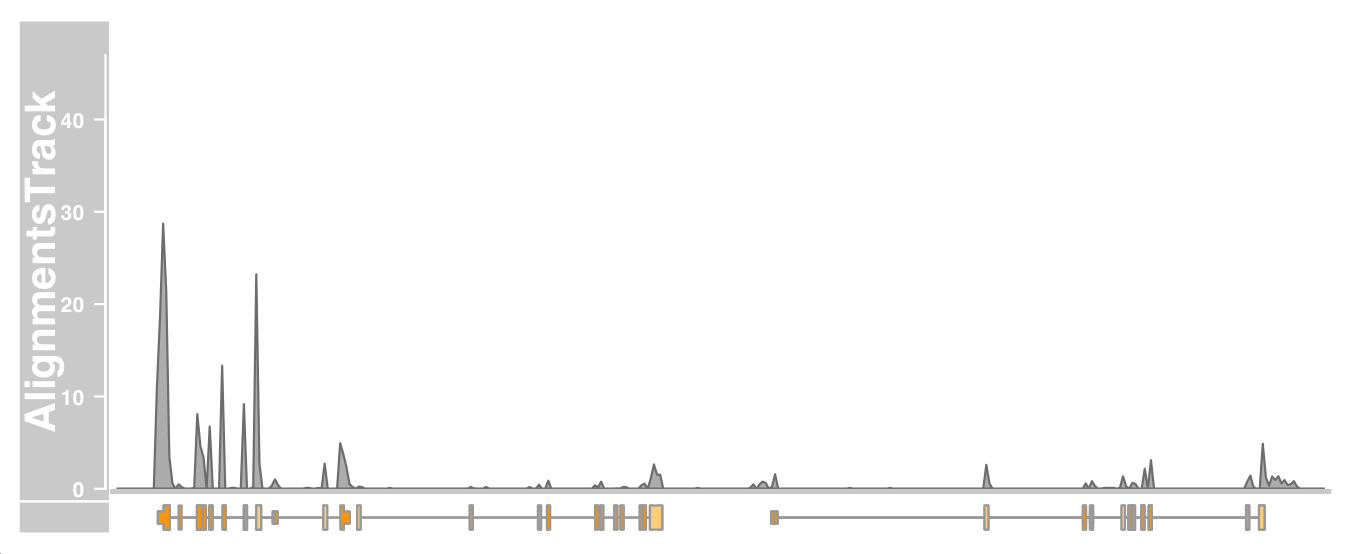
到目前为止,堆积 (pile-ups) 并不是特别有用,我们可以通过相应地设置类型显示参数来关闭它们。
1 plotTracks(c (alTrack, bmt), from = afrom, to = ato, chromosome = "chr12" , type = "coverage" )
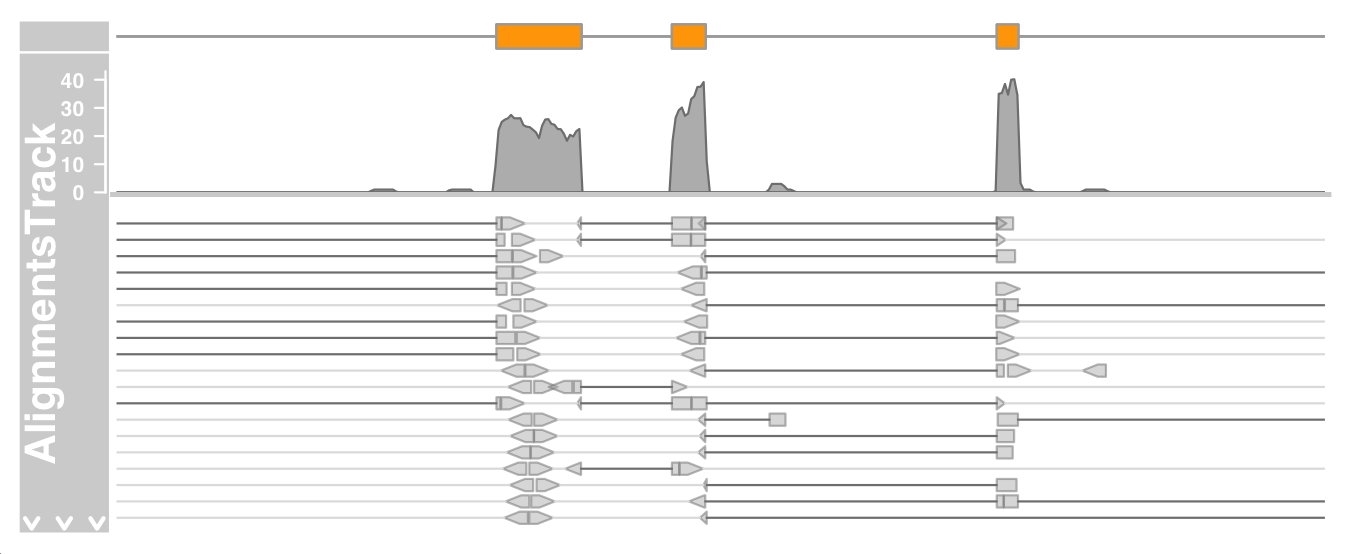
进一步放大以查看堆积部分的详细信息:
1 2 plotTracks(c (bmt, alTrack), from = afrom + 12700 , to = afrom + 15200 , chromosome = "chr12" )
单个reads的方向由箭头指示 亮灰色线连接 gaps 由连接的深灰色线显示
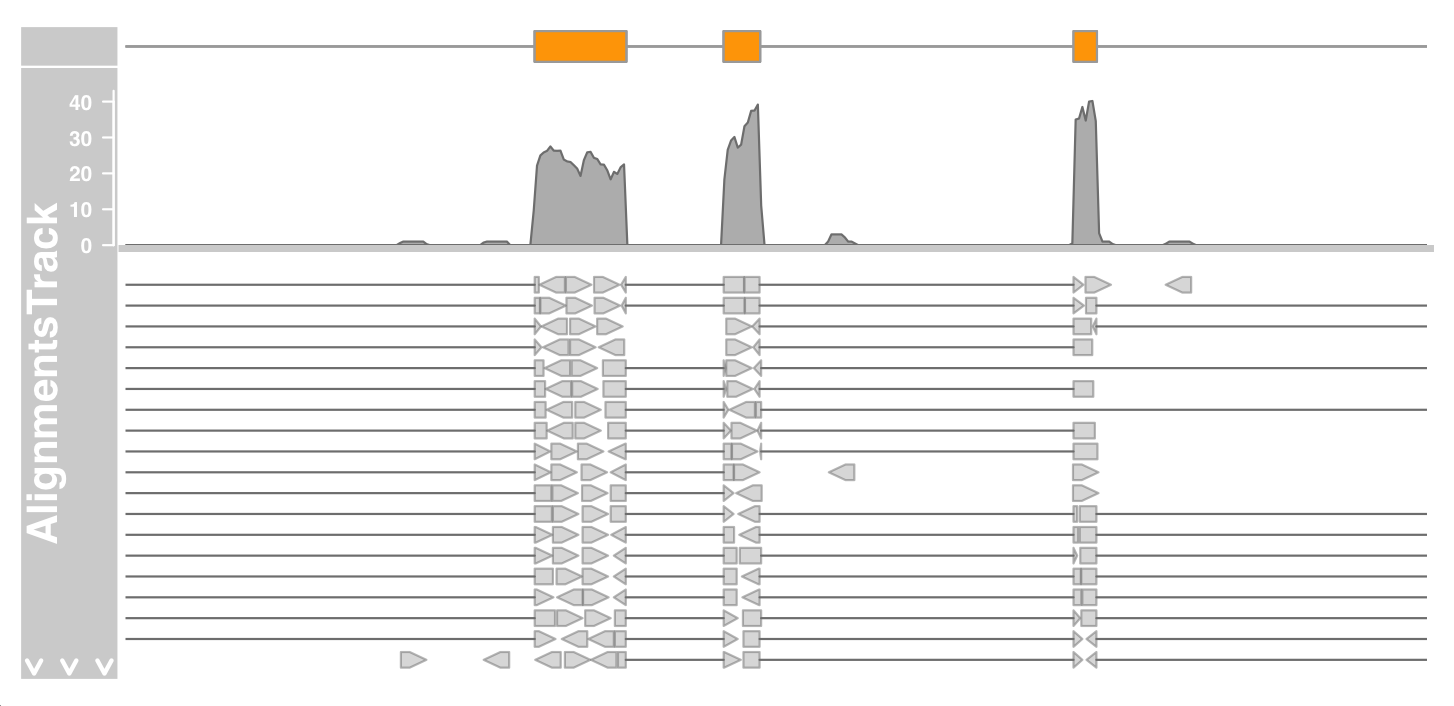
如前所述,我们可以通过在构造函数中设置 isPaired 参数 来控制数据应该被视为双端数据还是单端数据。
下面是我们在单端模式下查看同一文件中的数据
1 2 3 4 alTrack <- AlignmentsTrack(system.file(package = "Gviz" , "extdata" , "gapped.bam" ), isPaired = FALSE ) plotTracks(c (bmt, alTrack), from = afrom + 12700 , to = afrom + 15200 , chromosome = "chr12" )
DNA-seq
为了更好地展示 AlignmentsTrack 的序列变异特征,我们将加载不同的数据集,这次来自全基因组 DNA-Seq SNP calling 。
同样,参考基因组是 hg19,并且使用 Bowtie2 进行了比对。
我们需要指定 AlignmentsTrack 关于参考基因组(sequenceTrack )。
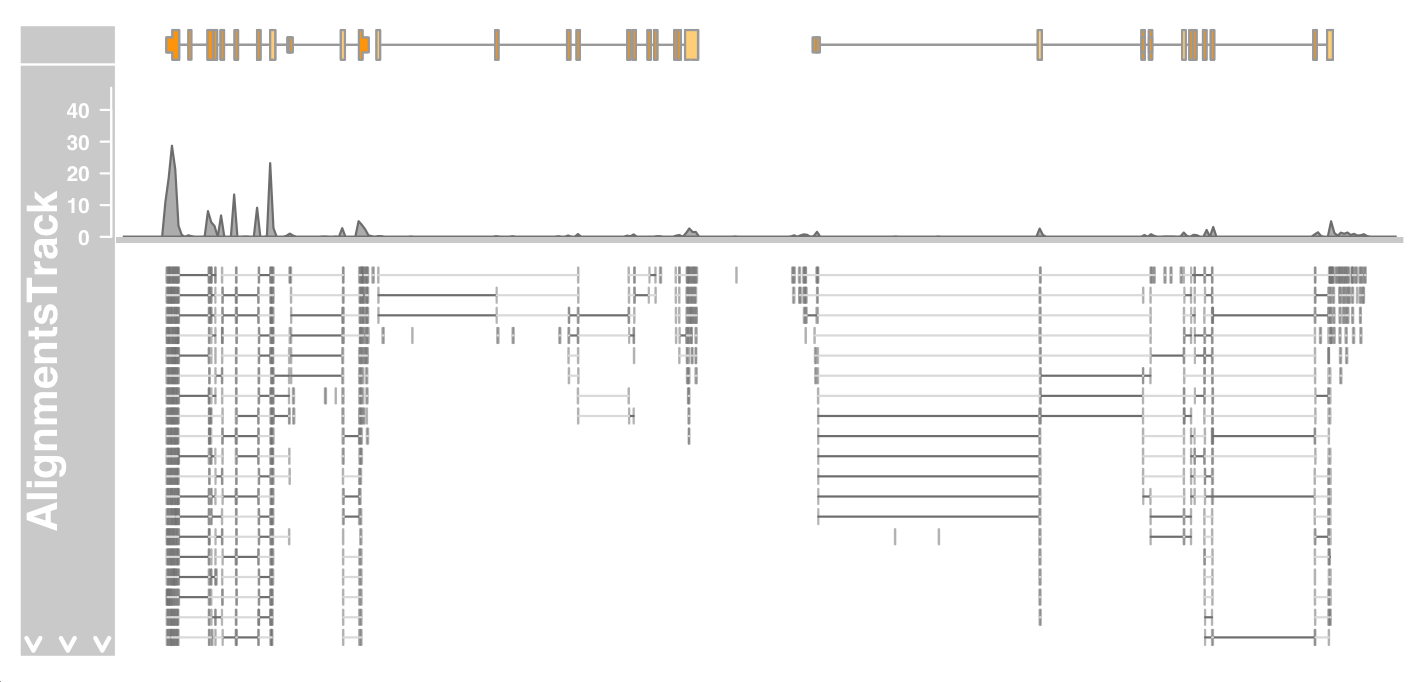
1 2 3 4 afrom <- 44945200 ato <- 44947200 alTrack <- AlignmentsTrack(system.file(package = "Gviz" ,"extdata" , "snps.bam" ), isPaired = TRUE ) plotTracks(c (alTrack, sTrack), chromosome = "chr21" , from = afrom,to = ato)
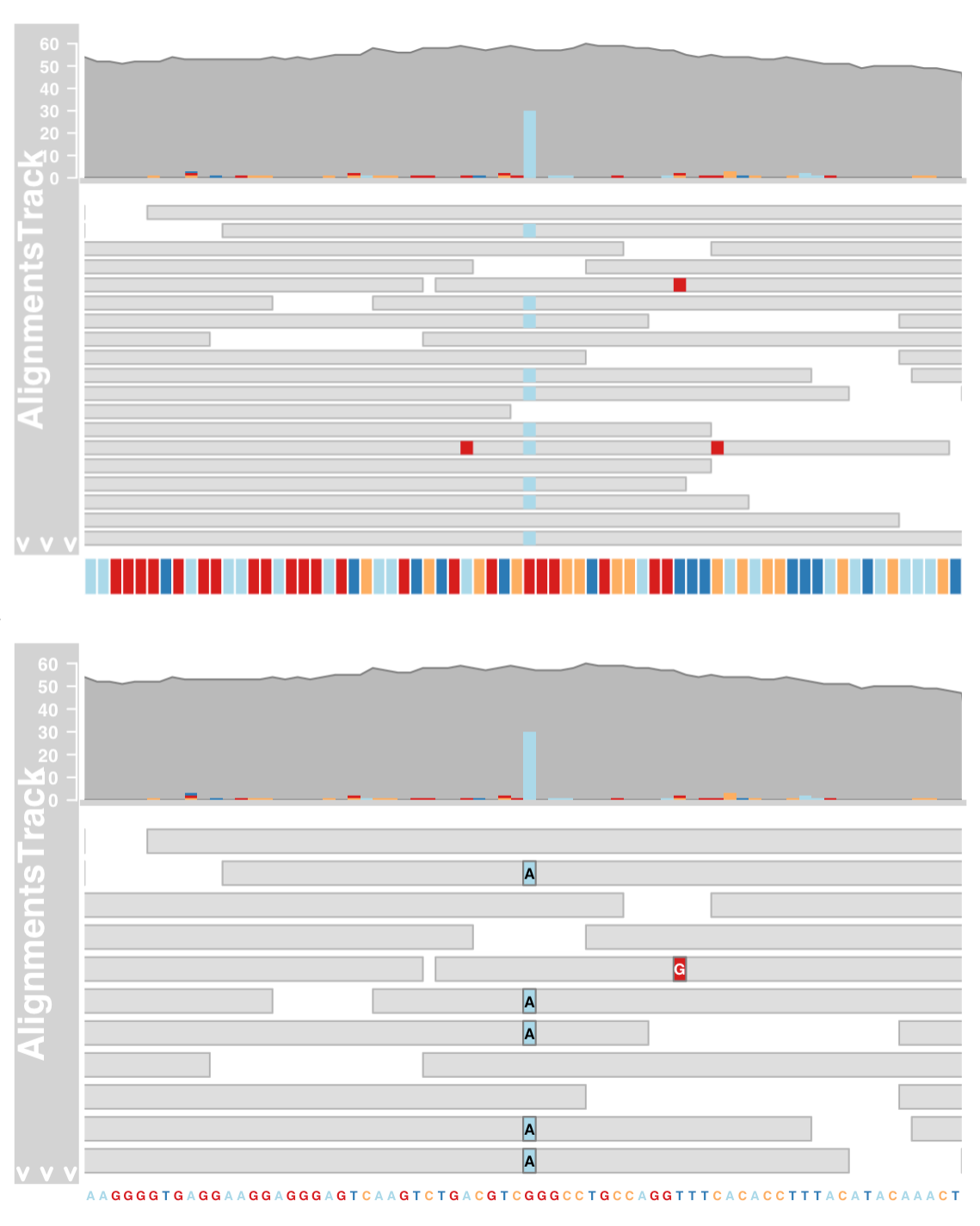
不匹配的碱基以叠加直方图的形式显示在堆积部分和覆盖图中的单个read上。
当放大到明显的杂合 SNP 位置之一时,可以揭示更多细节。
1 2 3 4 5 6 plotTracks(c (alTrack, sTrack), chromosome = "chr21" , from = 44946590 , to = 44946660 ) plotTracks(c (alTrack, sTrack), chromosome = "chr21" , from = 44946590 , to = 44946660 , cex = 0.5 , min.height = 8 )
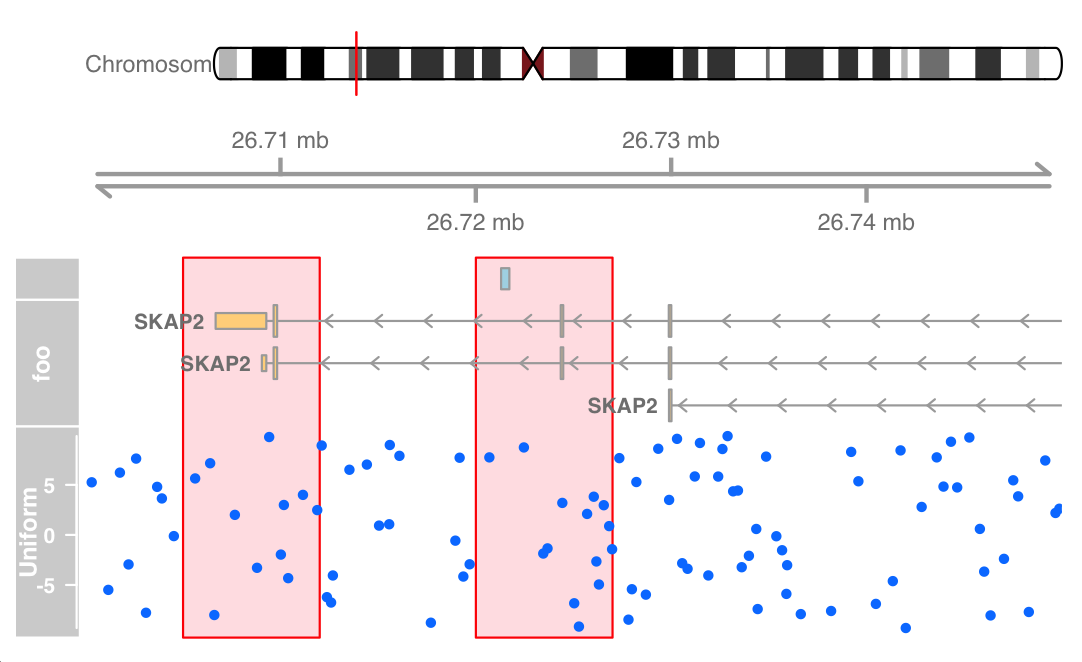
Track突出显示和叠加
突出显示
1 2 3 4 ht <- HighlightTrack(trackList = list (atrack, grtrack,dtrack), start = c (26705000 , 26720000 ), width = 7000 , chromosome = 7 ) plotTracks(list (itrack, gtrack, ht), from = lim[1 ], to = lim[2 ])
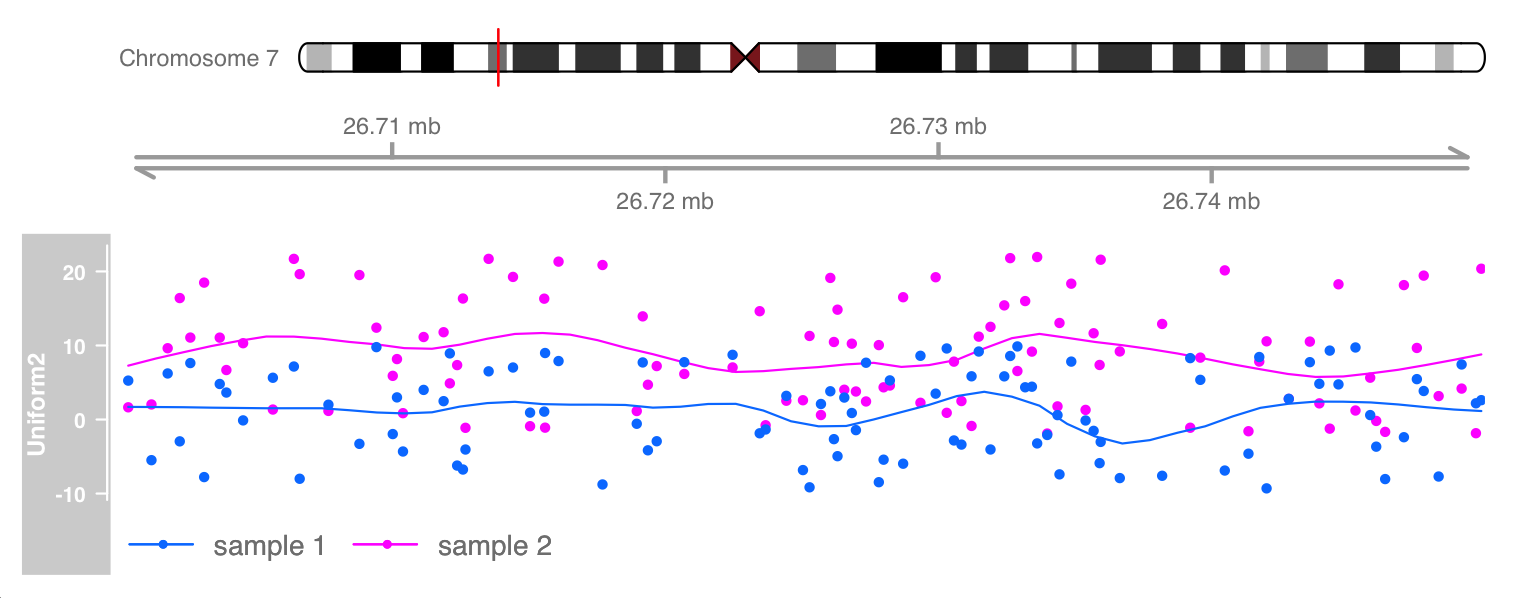
叠加
对于某些应用程序,在绘图的同一区域上叠加多个track 是有意义的。
为了举例,我们将生成第二个 DataTrack 对象并将其与第二章中的现有对象组合。
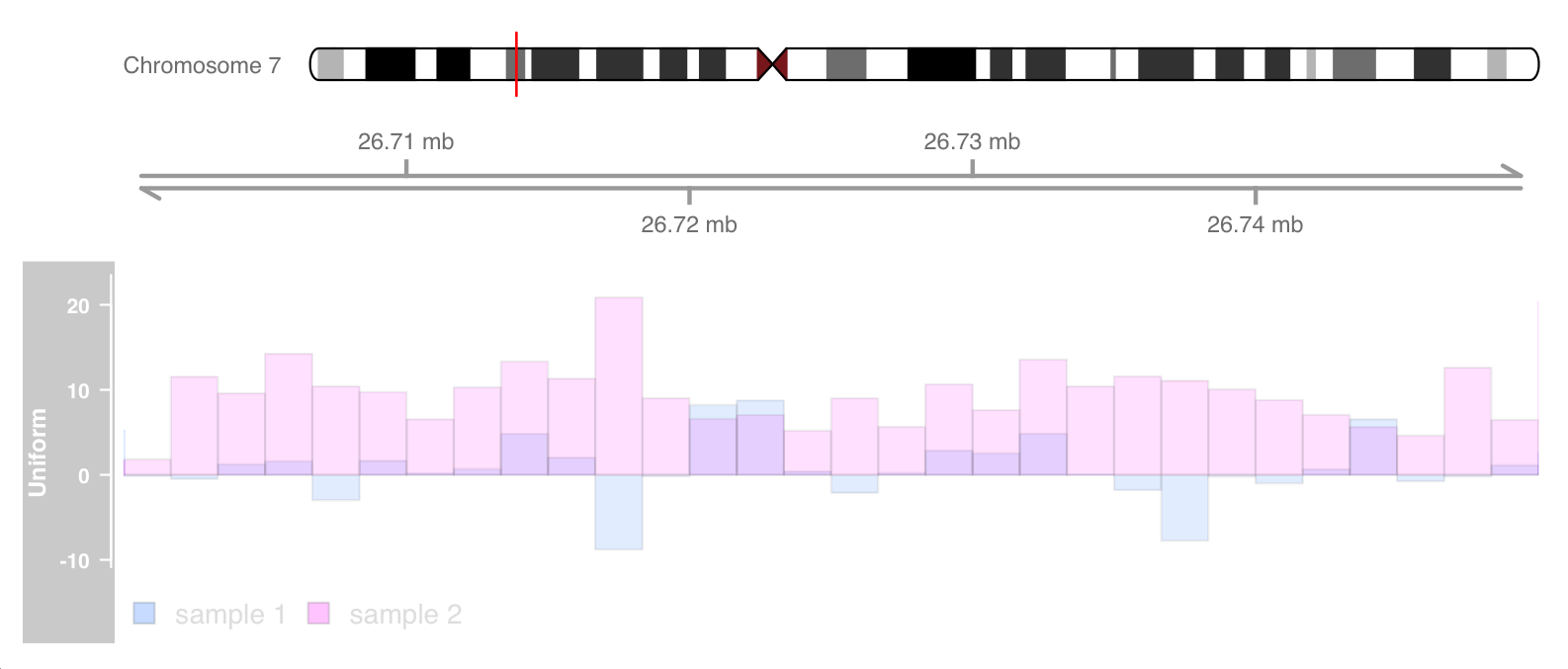
1 2 3 4 5 6 7 8 9 10 dat <- runif(100 , min = -2 , max = 22 ) dtrack2 <- DataTrack(data = dat, start = coords[-length (coords)], end = coords[-1 ], chromosome = chr, genome = gen, name = "Uniform2" , groups = factor("sample 2" ,levels = c ("sample 1" , "sample 2" )), legend = TRUE ) displayPars(dtrack) <- list (groups = factor("sample 1" ,levels = c ("sample 1" , "sample 2" )), legend = TRUE ) ot <- OverlayTrack(trackList = list (dtrack2, dtrack)) ylims <- extendrange(range (c (values(dtrack), values(dtrack2)))) plotTracks(list (itrack, gtrack, ot), from = lim[1 ], to = lim[2 ], ylim = ylims, type = c ("smooth" , "p" ))
在支持它的设备上,透明度混合 (Alpha blending ) 是一个有用的工具,可以从 track 覆盖中梳理出更多信息,至少在比较少量样本时是这样。
由此产生的透明度有效地消除了过度绘图的问题。以下示例仅在此小插图构建在具有透明度混合支持的系统上时才有效。
1 2 3 4 5 displayPars(dtrack) <- list (alpha.title = 1 , alpha = 0.5 ) displayPars(dtrack2) <- list (alpha.title = 1 , alpha = 0.5 ) ot <- OverlayTrack(trackList = list (dtrack, dtrack2)) plotTracks(list (itrack, gtrack, ot), from = lim[1 ], to = lim[2 ], ylim = ylims, type = c ("hist" ), window = 30 )
参考
Gviz http://www.bioconductor.org/packages/release/bioc/html/Gviz.html
https://github.com/ivanek/Gviz
Hahne, Florian, and Robert Ivanek. 2016. “Statistical Genomics: Methods and Protocols.” In, edited by Ewy Mathé and Sean Davis, 335–51. New York, NY: Springer New York. https://doi.org/10.1007/978-1-4939-3578-9_16 .
Katz, Yarden, Eric T. Wang, Jacob Silterra, Schraga Schwartz, Bang Wong, Helga Thorvaldsdóttir, James T. Robinson, Jill P. Mesirov, Edoardo M. Airoldi, and Christopher B. Burge. 2015. “Quantitative Visualization of Alternative Exon Expression from Rna-Seq Data.” Bioinformatics. https://doi.org/10.1093/bioinformatics/btv034 .
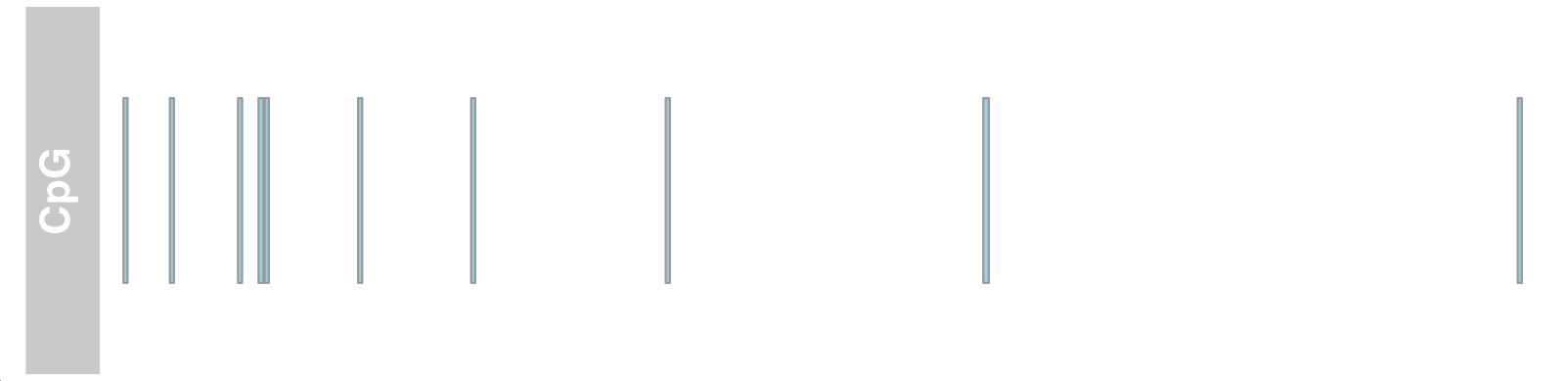


 微信
微信 支付宝
支付宝




