Hi-C辅助基因组组装原理|主流软件
导语
-
Hi-C是高通量染色体构象捕获(High-throughput Chromosome Conformation Capture, Hi-C)技术的简称,开发于2009年,最初用于捕获全基因组范围内所有的染色质内和染色质间的空间互作信息,目前已应用于基因表达的空间调控机制研究、构建染色体水平参考基因组、构建单体型图谱等。
-
Hi-C技术源于染色体构象捕获(Chromosome Conformation Capture, 3C)技术,利用高通量测序技术,结合生物信息分析方法,研究全基因组范围内整个染色质DNA在空间位置上的关系,获得高分辨率的染色质三维结构信息。Hi-C技术不仅可以研究染色体片段之间的相互作用,建立基因组折叠模型,还可以应用于基因组组装、单体型图谱构建、辅助宏基因组组装等,并可以与RNA-Seq、ChIP-Seq等数据进行联合分析,从基因调控网络和表观遗传网络来阐述生物体性状形成的相关机制。
3C,4C,5C以及HiC测序技术
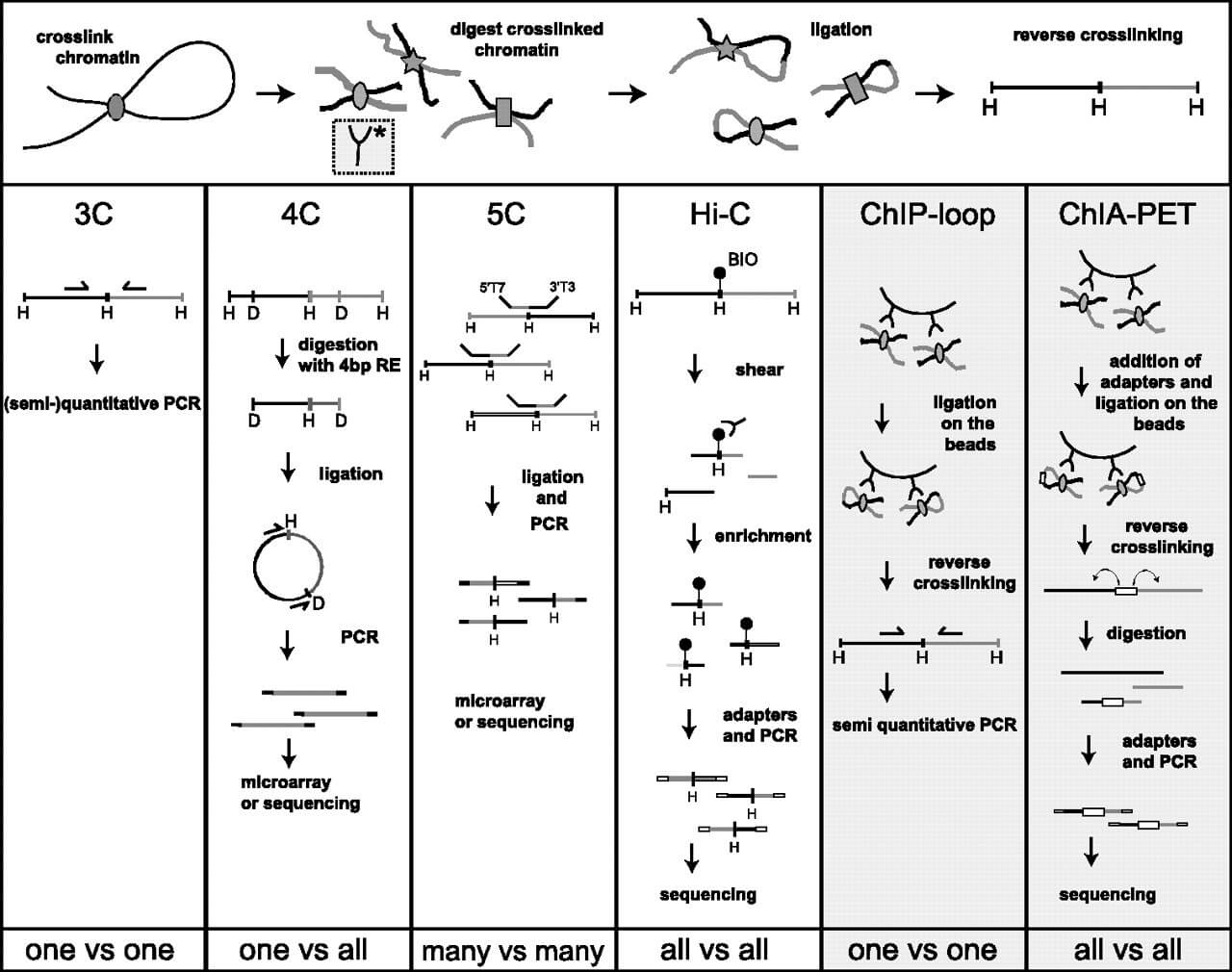
3C
3C,可以验证1个点与1个点的相互作用,每1对相互作用需要1对引物
4C
4C技术,可以验证1个点与多个点的相互作用,因为根据这1个点设计,关键步骤是成环。
5C
HiC
Hi-C,获得all-to-all的互作关系
- 具体如下图介绍:
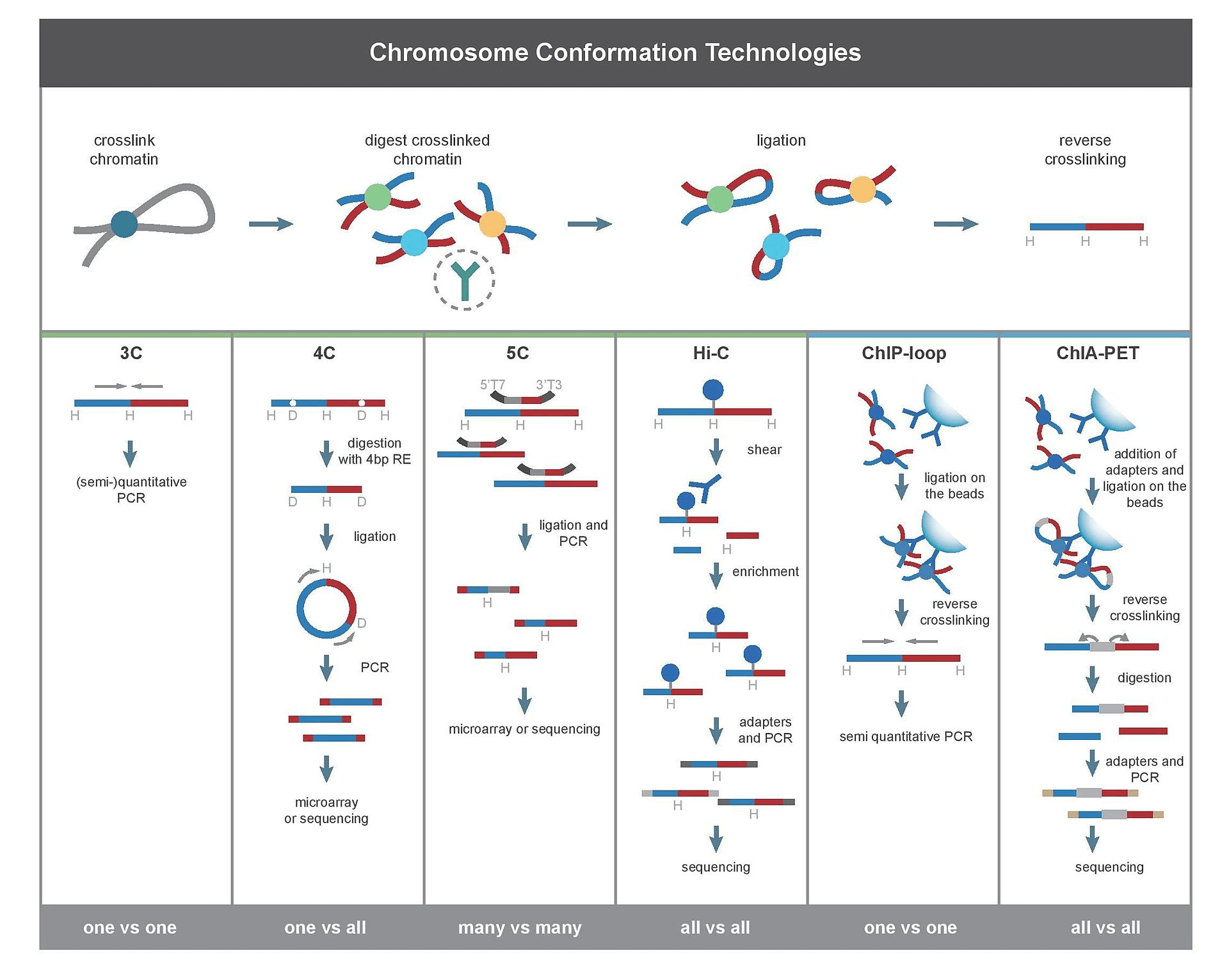
Hi-C辅助组装实验流程
利用甲醛对样本进行交联,质检合格后使用限制性内切酶(如MboI等)进行酶切,酶切片段经生物素标记、平末端连接、DNA纯化提取,超声打断后钓取含有生物素的片段,进行建库测序。
随后,对原始下机数据进行质控,并将质控截取后的Clean reads与参考基因组比对,获得用于互作分析的Valid reads。由于Hi-C文库的构建具有一定的复杂性,在实际的项目执行过程中,会先通过对小规模的测序数据进行评估,以检测所构建文库的质量。小数据评估合格后,启动大数据的上机测序,以保证测序数据的质量。
Hi-C技术的大致流程
-
通过甲醛交联固定,将细胞内由蛋白质介导的空间上邻近的染色质片段进行共价连接。
-
限制性内切酶进行酶切
-
使用生物素标记末端标记
-
将连接的DNA纯化后超声打断,并用生物素亲和层析,将生物素化的DNA片段分离,加上接头进行高通量测序
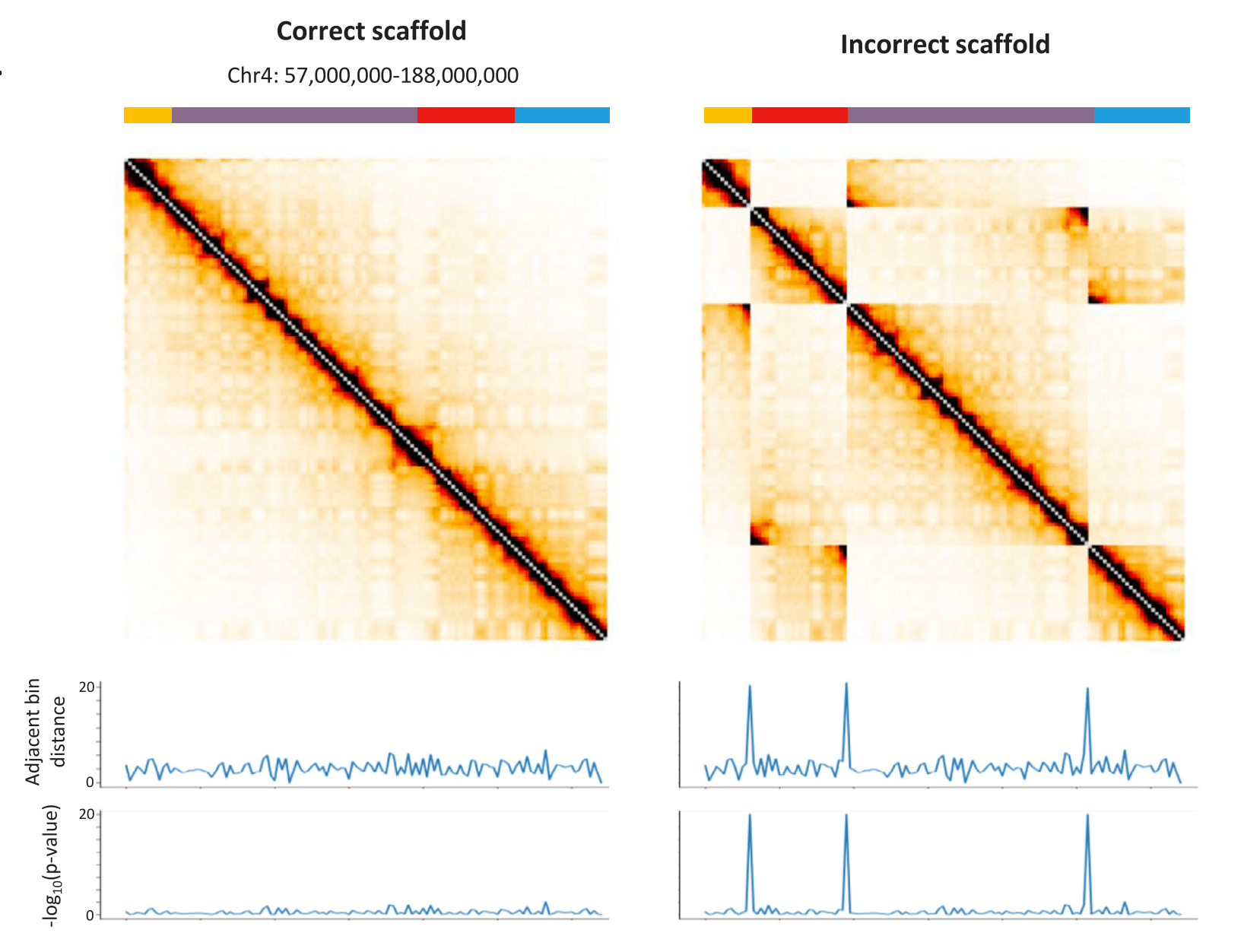
Hi-C互作三大规律
1.染色体内互作富集
2.互作随距离衰减
3.局部互作平滑
可以通过以上三个规律来判断组装的好坏
Hi-C建库测序流程
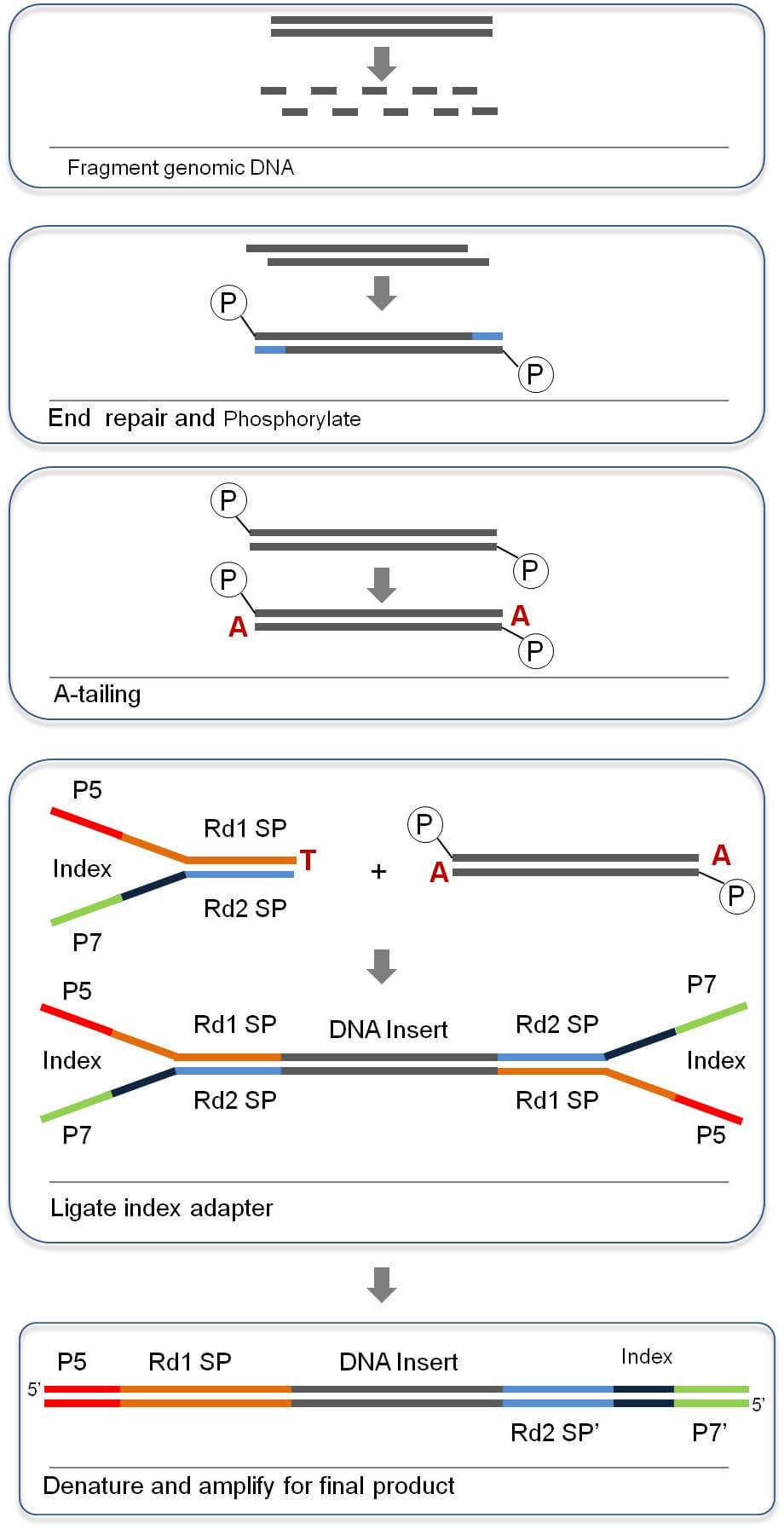
-
DNA样品检测
(1) 琼脂糖凝胶电泳分析DNA降解程度以及是否有污染
(2) Nanodrop检测DNA的纯度(OD260/280比值)
(3) Qubit对DNA浓度进行精确定量 -
文库构建
检测合格的DNA样品通过Covaris超声波破碎仪随机打断,经末端修复、加A尾、加测序接头、纯化、PCR扩增等步骤完成整个文库制备。文库构建原理图如下: -
库检
(1) Agilent 2100检测文库DNA片段的完整性及插入片段大小。
(2) QPCR及QPCR检测文库有效浓度。
检测合格后进行下一步上机测序。 -
上机测序
库检合格后,把不同文库按照有效浓度及目标下机数据量的需求pooling后进行Illumina测序。
Hi-C组装软件
在组装基因组时,使用二代或三代数据组装到contigs后,下一步就是将contig提升到染色体水平。利用HiC数据目前常见的组装软件有下面几个:
HiRise: 2015年后的GitHub就不再更新
LACHESIS: 发表在NBT,2017年后不再更新
SALSA: 发表在BMC genomics, 仍在更新中
3D-DNA: 发表在science,仍在更新中
ALLHiC: 发表在Nature Plants, 用于解决植物多倍体组装问题
HiC-Pro:发表在FGenome Biology
LACHESIS
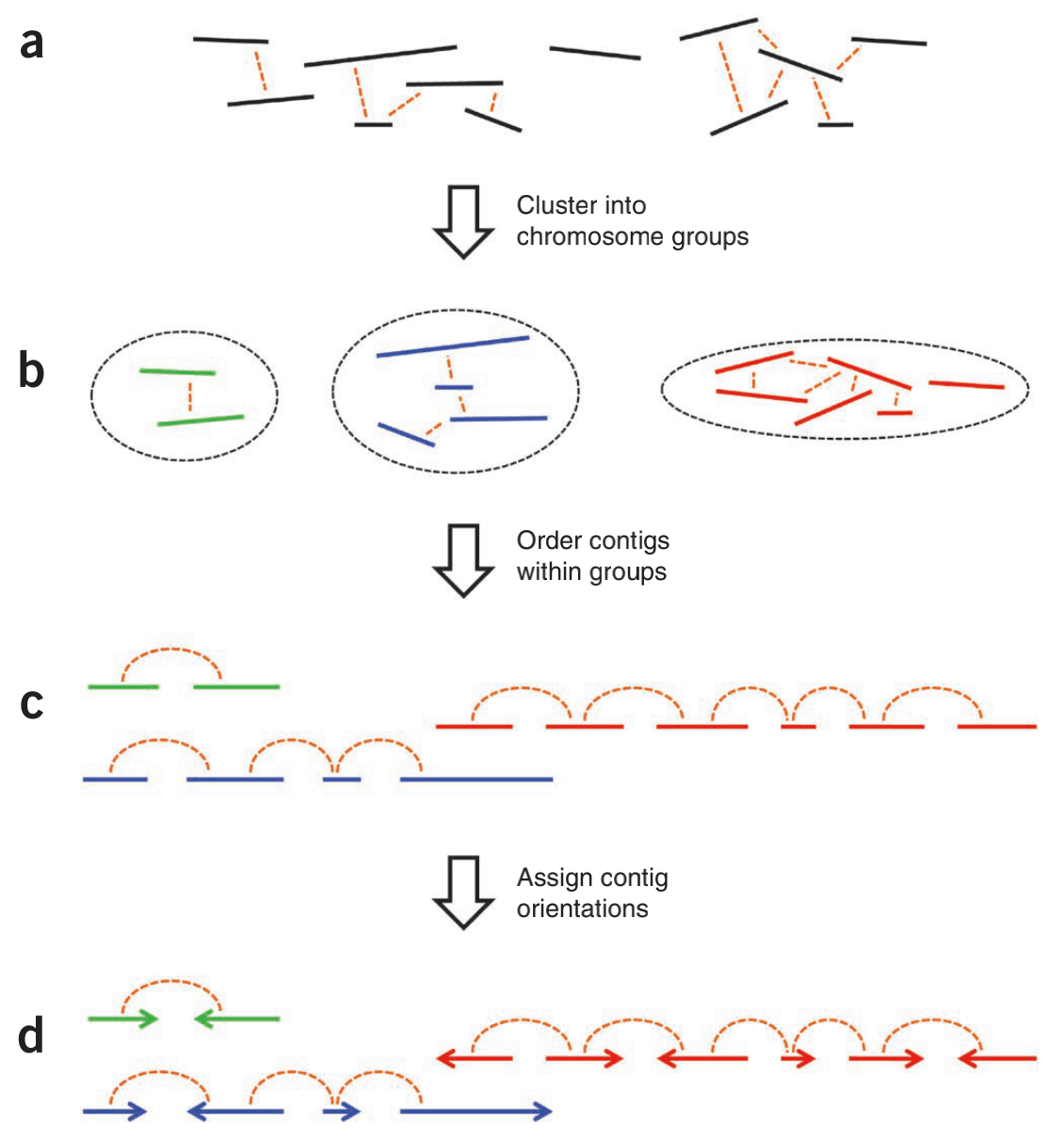
-
输入包括一组来自草稿装配的contigs (or scaffolds) 和一组全基因组染色质相互作用数据,例如Hi-C links。
-
与不同染色体上的contigs相比,同一染色体上的contigs之间往往有更多的Hi-C links。LACHESIS利用这一点将contigs聚集成与个体染色体基本一致的群体。
-
在一条染色体内,近在咫尺的contigs往往比相距遥远的contigs有更多的联系。LACHESIS利用这一点来排列每个染色体组内的contigs。
-
最后,LACHESIS利用相邻contigs之间连接的精确位置来预测每个contigs的相对方向。
HiC-Pro
HiC-Pro是一款高效的Hi-C数据分析软件,提供了从原始数据到归一化之后的HI-C图谱构建的完整功能,运行效率高,用法简便。
完整的pipeline如下图所示:
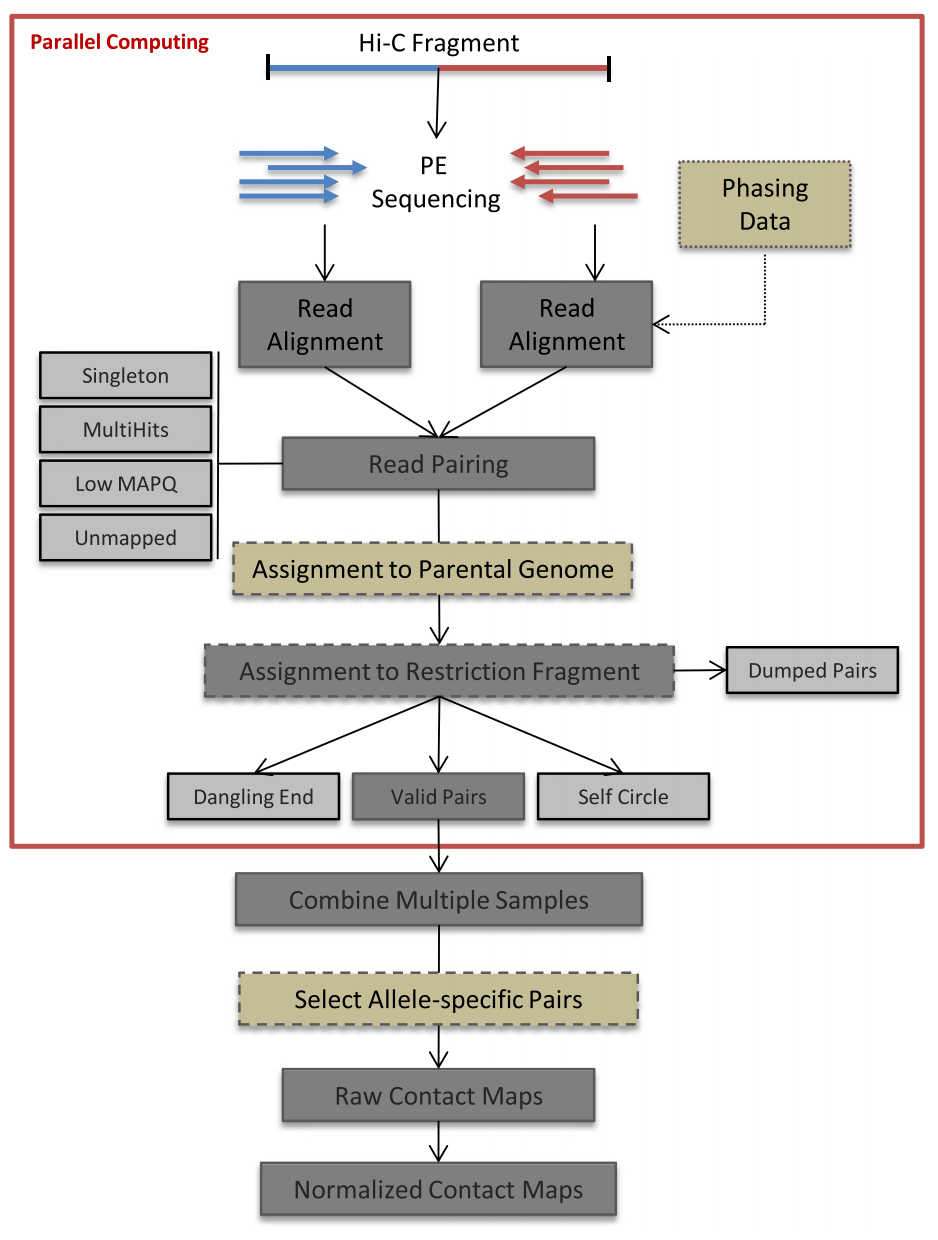
其具体上机使用方法可以参考链接
序列比对
筛选Valid pairs
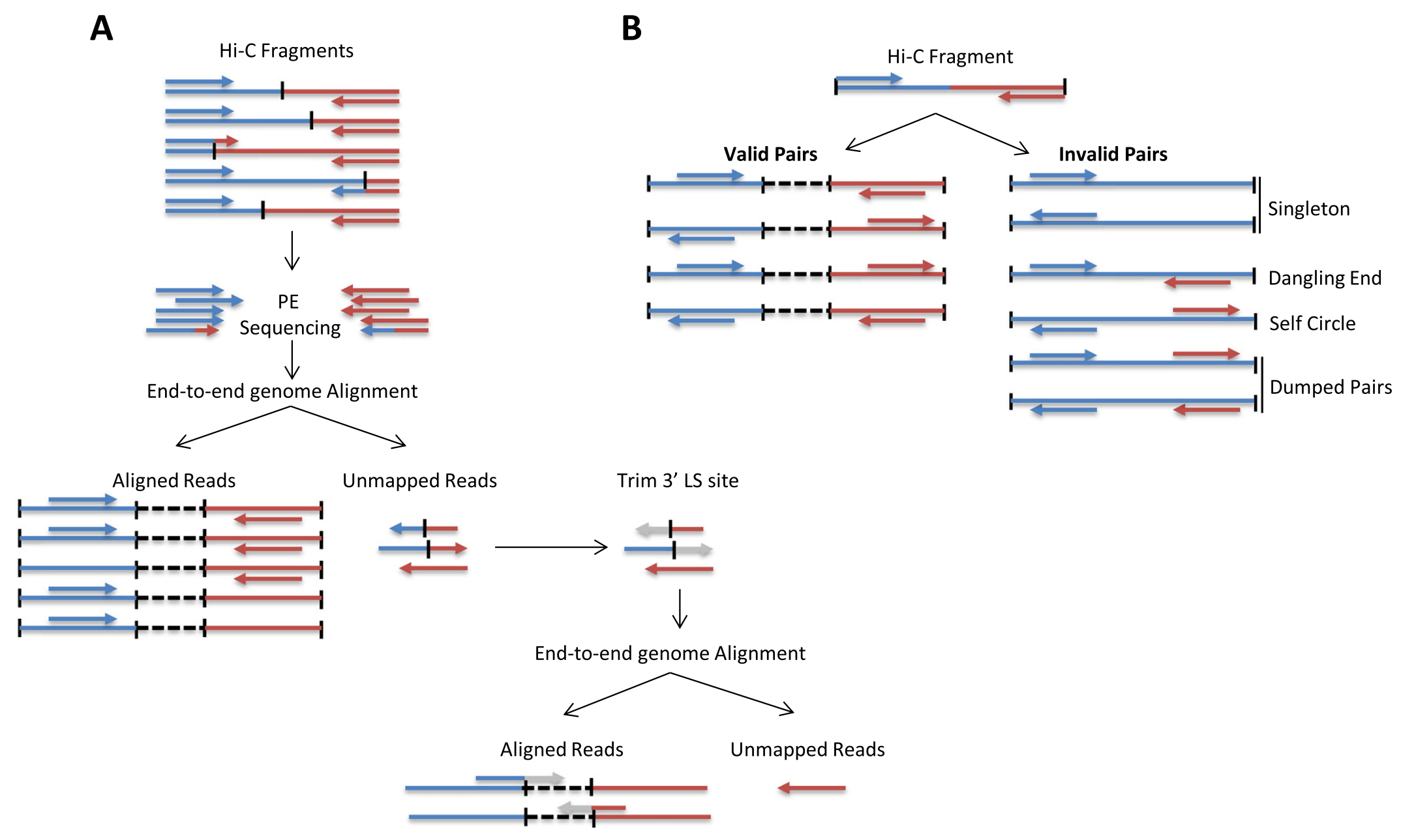
构建原始Hi-C图谱
根据指定的分辨率,统计两个bin区域内valid pairs的数目, 去除PCR重复之后,构建原始的交互矩阵。
归一化
不同区域GC含量,mapping概率等系统误差都使得原始的交互矩阵不能够有效代表染色质交互信息, 所以需要进行归一化。采用了一种迭代校正的归一化算法对原始的交互矩阵进行归一化,矫正系统误差。
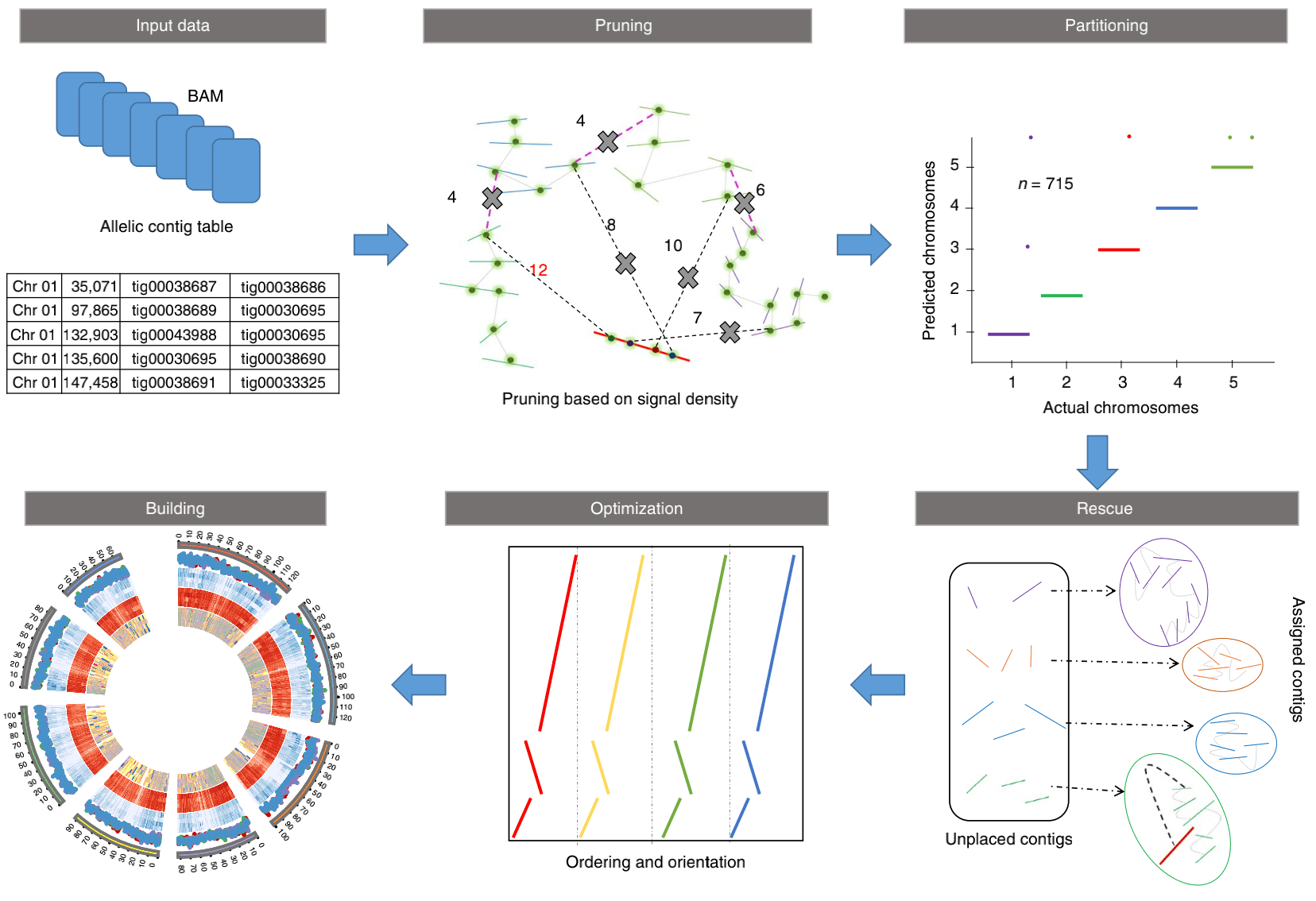
ALLHiC
ALLHiC一共分为五步:pruning, partition, rescue, optimization, building
-
prune 步骤去除了等位基因之间的联系,因此同源染色体更易于单独分离。
-
partition 功能将修剪的bam文件作为输入,并根据Hi-C建议的链接对链接的contigs进行聚类,大概是沿着相同同源染色体在预设数量的分区中进行。
-
rescue 功能从原始未修剪的bam文件中搜索分区步骤中不涉及的contigs,并根据Hi-C信号密度将它们分配给特定的群集。
-
optimize 步骤采用每个分区,并优化所有contigs的顺序和方向。
-
build 步骤通过连接contigs来重建每个染色体
如下图所示:
 ]
]
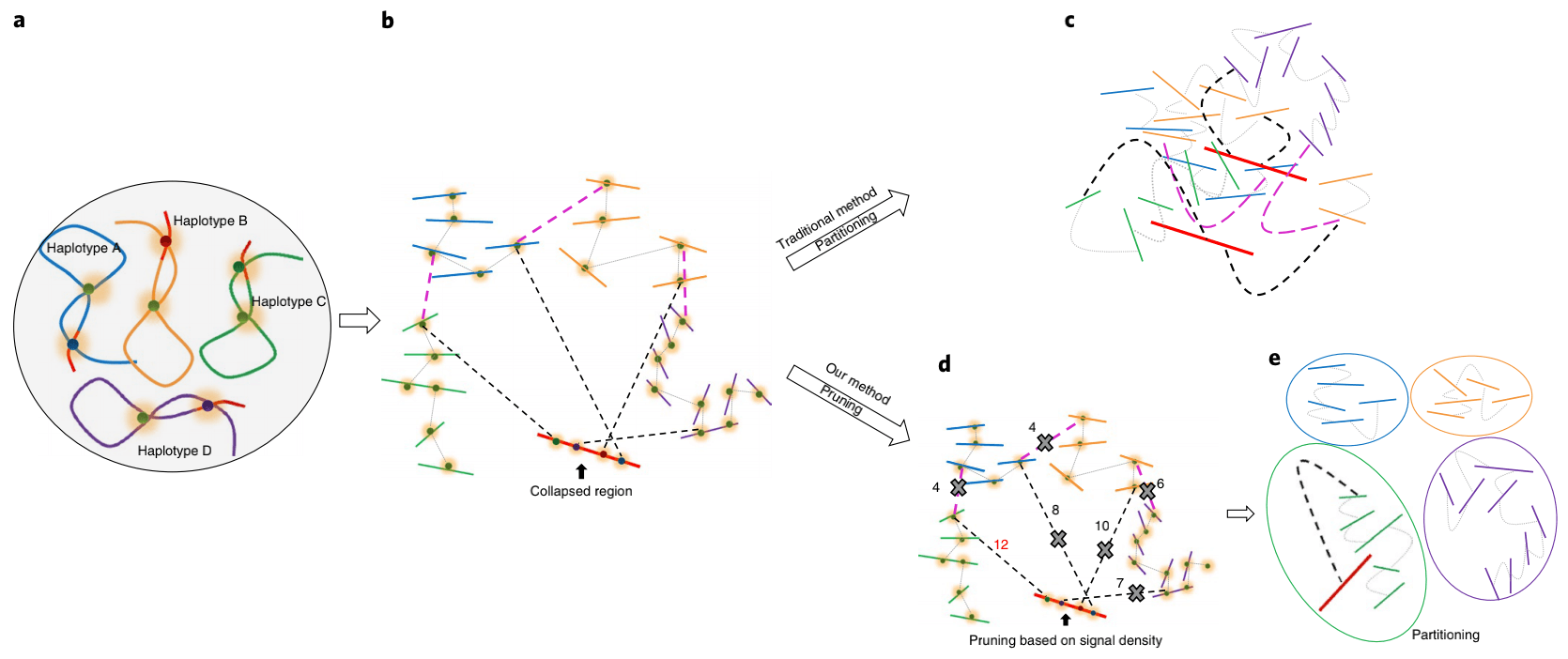
Explanation of Prune
-
同源四倍体基因组的示意图。四个同源染色体显示为不同的颜色(分别为蓝色,橙色,绿色和紫色)。染色体中的红色区域表示具有高度相似性的序列。
-
检测自身四倍体基因组中的Hi-C信号。黑色虚线表示折叠区域和未折叠区域contigs之间的Hi-C信号。粉色虚线表示单体型Hi-C链接,灰色虚线表示单体型Hi-C链接。在组装过程中,红色区域会因高度的序列相似性而崩溃;同时,如果其他区域之间存在大量差异,则会将它们分为不同的contigs。由于塌陷区域与来自不同单倍型的contigs在物理上相关,因此将在塌陷区域与所有其他未塌陷的contigs之间检测到Hi-C信号。
-
传统的Hi-C脚手架方法将检测来自不同单倍型和折叠区域的contigs中的信号,并将所有序列聚在一起。
-
修剪Hi-C信号:1-去除等位基因区域之间的信号;2-仅在折叠区域和未折叠contigs之间保留最强的信号。
-
基于修剪的Hi-C信息进行分区。理想情况下,根据修剪结果将contigs分为不同的组。
参考文献
[1] Burton, J., Adey, A., Patwardhan, R. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol 31, 1119–1125 (2013).
[2] Servant N, Varoquaux N, Lajoie B R, et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing[J]. Genome biology, 2015, 16(1): 1-11.
[3] Lieberman-Aiden E, Van Berkum N L, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome[J]. science, 2009, 326(5950): 289-293.
[4] Ghurye J, Rhie A, Walenz B P, et al. Integrating Hi-C links with assembly graphs for chromosome-scale assembly[J]. PLoS computational biology, 2019, 15(8): e1007273.
[5] Dudchenko O, Batra S S, Omer A D, et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds[J]. Science, 2017, 356(6333): 92-95.
[6] Zhang X, Zhang S, Zhao Q, et al. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data[J]. Nature plants, 2019, 5(8): 833-845.
[7] Durand N C, Shamim M S, Machol I, et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments[J]. Cell systems, 2016, 3(1): 95-98.
[8] Wu S, Turner K M, Nguyen N, et al. Circular ecDNA promotes accessible chromatin and high oncogene expression[J]. Nature, 2019, 575(7784): 699-703.
[9] Oddes S, Zelig A, Kaplan N. Three invariant Hi-C interaction patterns: applications to genome assembly[J]. Methods, 2018, 142: 89-99.
[10] Zhang, J. Zhang, X. Tang, H. Zhang, Q. et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nature Genetics, doi:10.1038/s41588-018-0237-2 (2018).
 微信
微信 支付宝
支付宝











