物种内共线性分析——思路以及踩坑总结(二)
物种内共线性分析(MCScanX+BLAST+TBtools)
数据要求:做物种内共线性分析的话主要需要的是
全基因组序列、cds或pep序列、gff3/gtf序列三者缺一不可。
上一节下载好了cds序列以及gff3序列文件,以此为例
(数据可在Phyzome下载,也可以在服务器上在线下载)
软件要求:MCScanX、blast、TBtools(JCVI)
物种内blast
物种内blast 使用cds或pep序列进行自我比对,结果*.blast格式得到此结果(这一步耗时最长,可以使用TBtools一键完成,有服务器的同学可以使用服务器运行)
blast构建索引 | makeblastdb
1 | makeblastdb -in Zmarina_324_v2.2.cds.fa -dbtype nucl -out Zmarina.db |
参数说明:
-in 后接输入文件,你要格式化的fasta序列
-dbtype 后接序列类型,nucl为核酸,prot为蛋白
-out 后接数据库名,自定义,后续blast+要用到的-db的参数
-logfile 日志文件,如果没有默认输出到屏幕
比对核酸数据库(blastn)
如果下载的cds序列
1 | blastn -query Zmarina_324_v2.2.cds.fa -db Zmarina.db -out Zmarina.blast -evalue 1e-10 -num_threads 10 -outfmt 6 -num_alignments 5 |
比对蛋白数据库(blastp)
如果下载为pep序列
1 | blastp -query Zmarina_324_v2.2.pep.fa -db Zmarina.db -out Zmarina.blast -evalue 1e-10 -num_threads 10 -outfmt 6 -num_alignments 5 |
参数说明:
-query: 输入文件路径及文件名
-out:输出文件路径及文件名
-db:格式化了的数据库路径及数据库名
-outfmt:输出文件格式,总共有12种格式,6是tabular格式对应BLAST的m8格式
-evalue:设置输出结果的e-value值
-num_threads:线程数
-num_alignments: 设置每个query保留多少条匹配结果
gff序列简化
已知gff序列分成许多行,其实我们只需要四行,所以需要将这四行提取出来得到简化后的gff文件
简化的步骤可用脚本获得如下:
1 | ##Phytozome GFF3文件处理 |
MCScanX
命令行格式:MCScanX + 名称(这里需要注意的是前面得到的简化后的gff文件以及blast之后的结果文件;命名须一致)
比如得到的blast结果为zm.blast,简化为四列后的gff文件为zm.gff
使用命令MCScanX zm即可得出共线性结果。
1 | $ ls -lh |
运行此软件即可得出结果文件:
名称.html,
名称.collinearity, (包含了共线性分析的结果)
名称.tandem(串联重复)
在这一步遇到了问题是一直出不来结果的原因:
上面gff格式的问题tab改为空格等原因导致软件跑不出来共线性。可以通过文本软件检查一下格式是否准确
可视化circos
在这里可以用的有TBtools、circos、JCVI等软件进行可视化其中需要配置很多的文件进行分析出图
准备好简化后的四列gff文件;*.blast文件
以及MCScanX分析得出的.collinearity和.tandem进一步分析步骤
这里以TBtools为例进行说明:
整体流程如下~
- 用到 Advanced Circos 模块
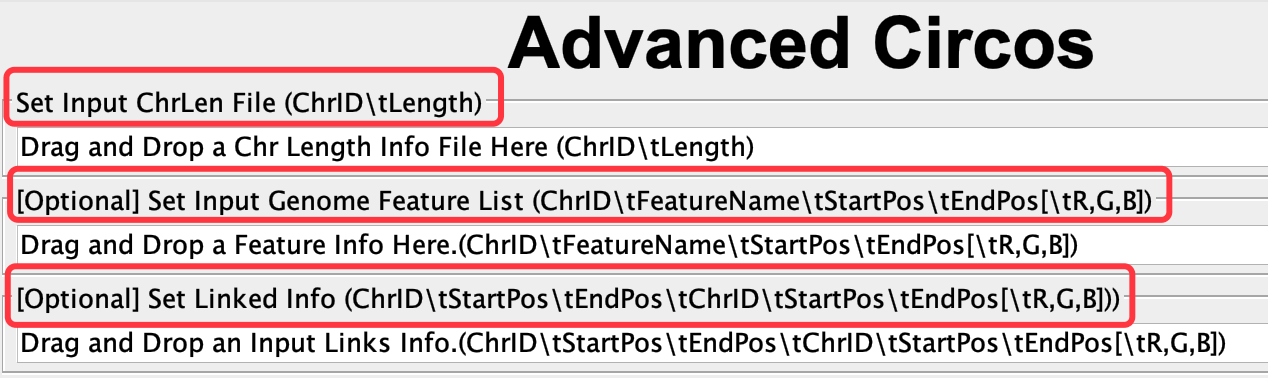
需要准备的文件如下:
-
染色体长度文件:
用到 fasta stat 模块, 将基因组的文件输入进去,输出整个染色体长度的文件;提取染色体的长度信息,保存为文本文件,ChrLen.txt ( Advanced Circos 模块 需要的文件1) -
基因组内的共线性:
将共线性分析结果,转换成GenePairTable(模块Text Merge for MCScanX;输入前面得到的.collinearity文件,Merge Mode选择Collinear输出txt文件命名为GenePair.tab.txt),之后需要继续转换为LinkedRegion文件(模块Text Transformat for Micro-Synteny View;Input File Format为GenePair,输出命名为LickedRegion.tab.txt)
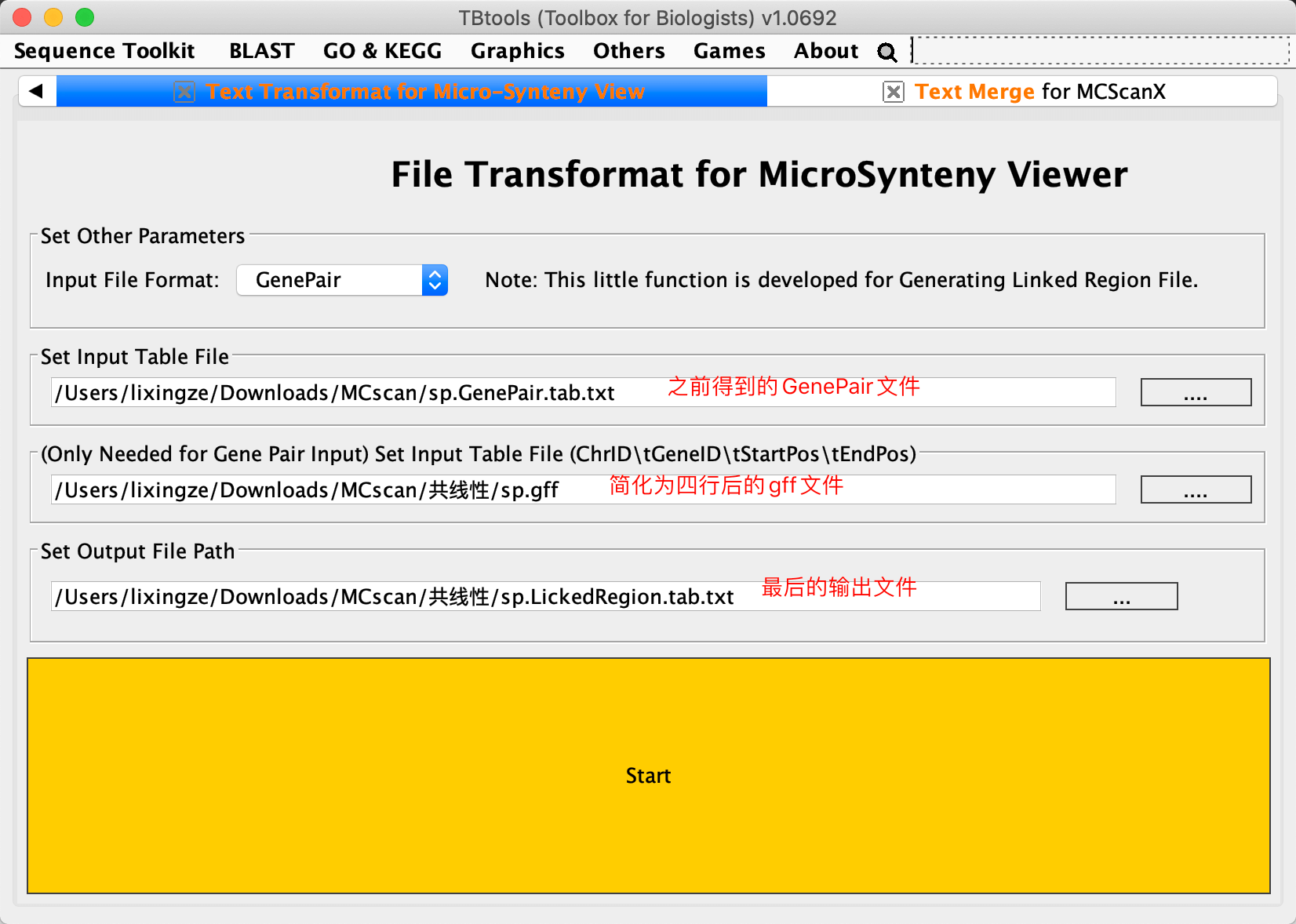
这一步做完就得到了他们的共线性关系
需要的是展示WRKY基因家族内部参与的复制事件,所以与WRKY·ID相关的连接线应该被高亮出来。或者我们直接补充一些高亮的线进去就可以了
直接使用TBtools的文本区块提取工具【Text Block Extract】
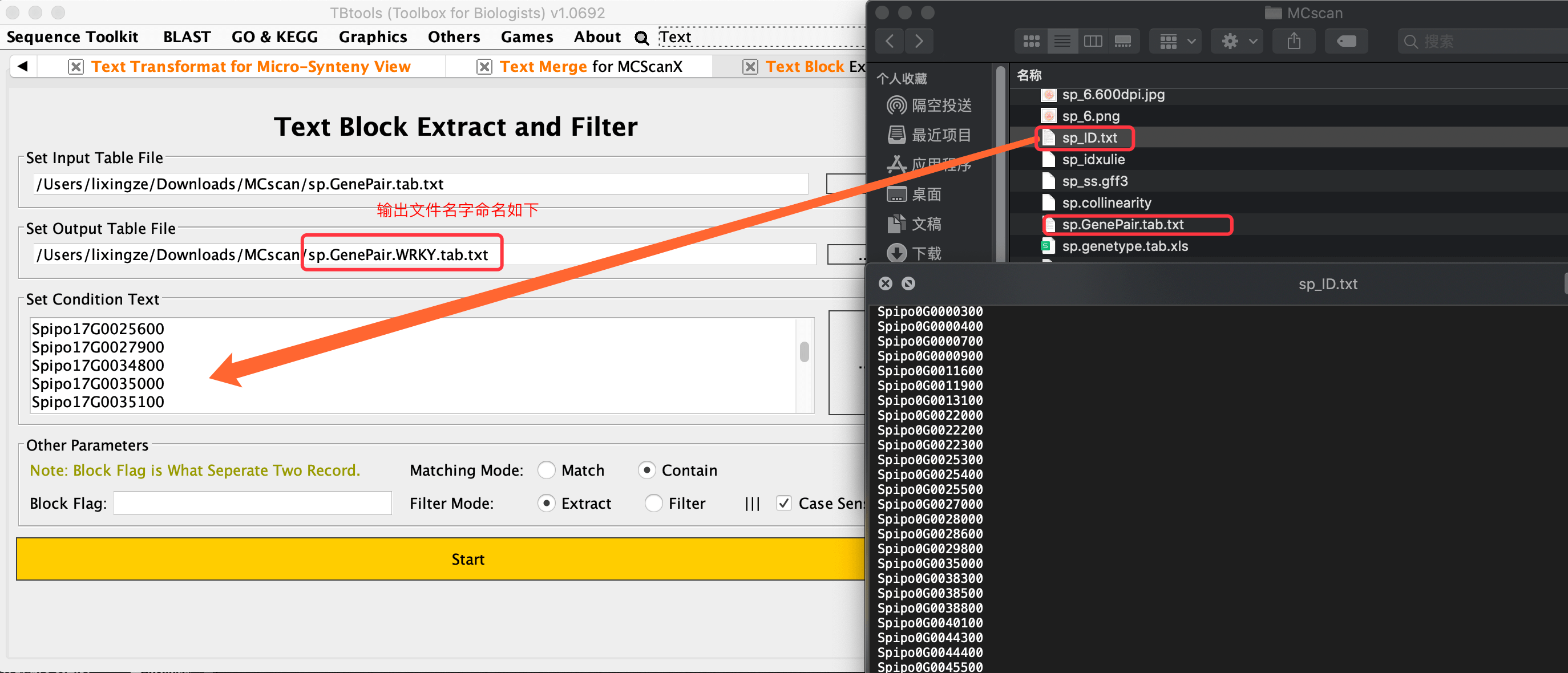
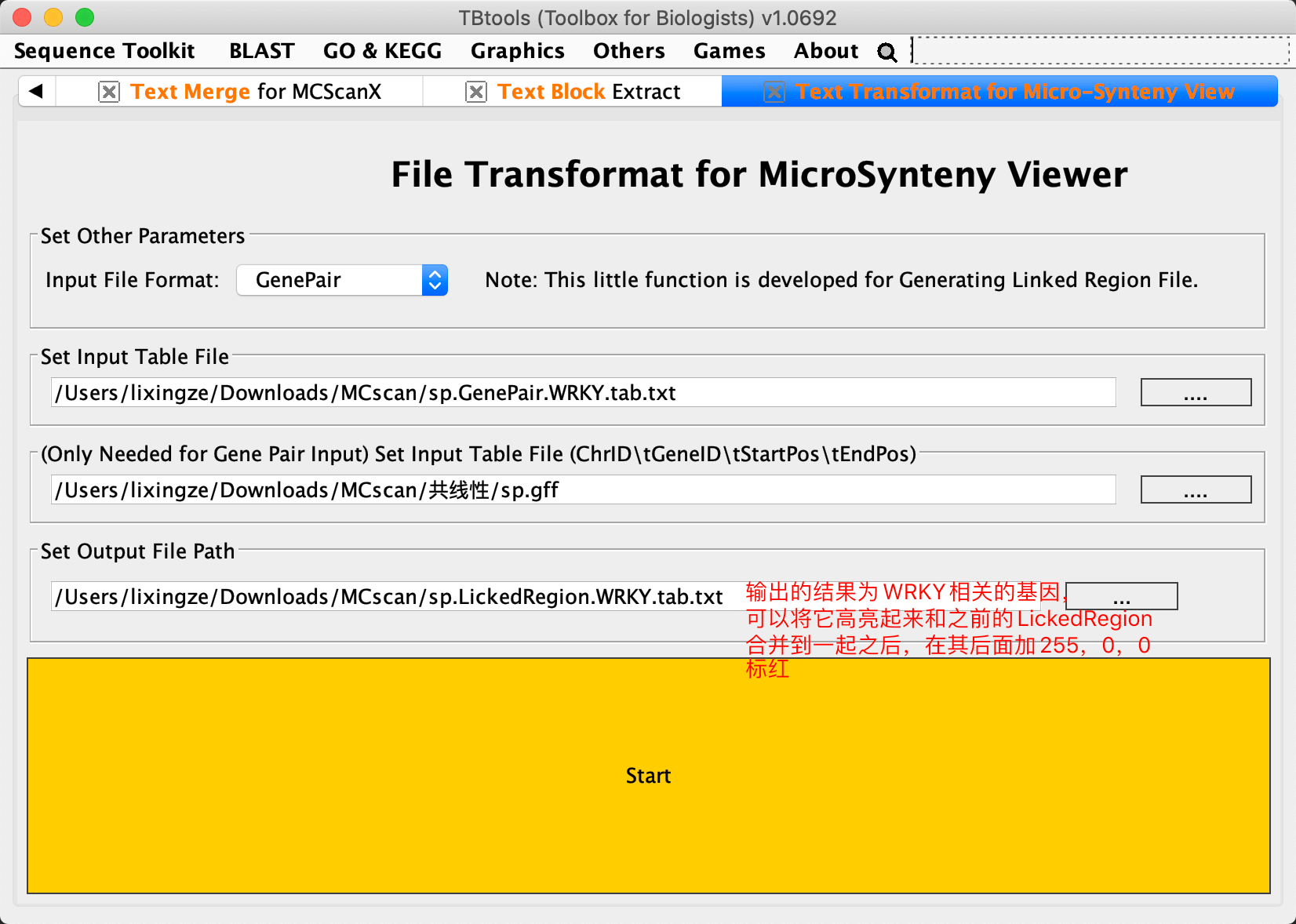
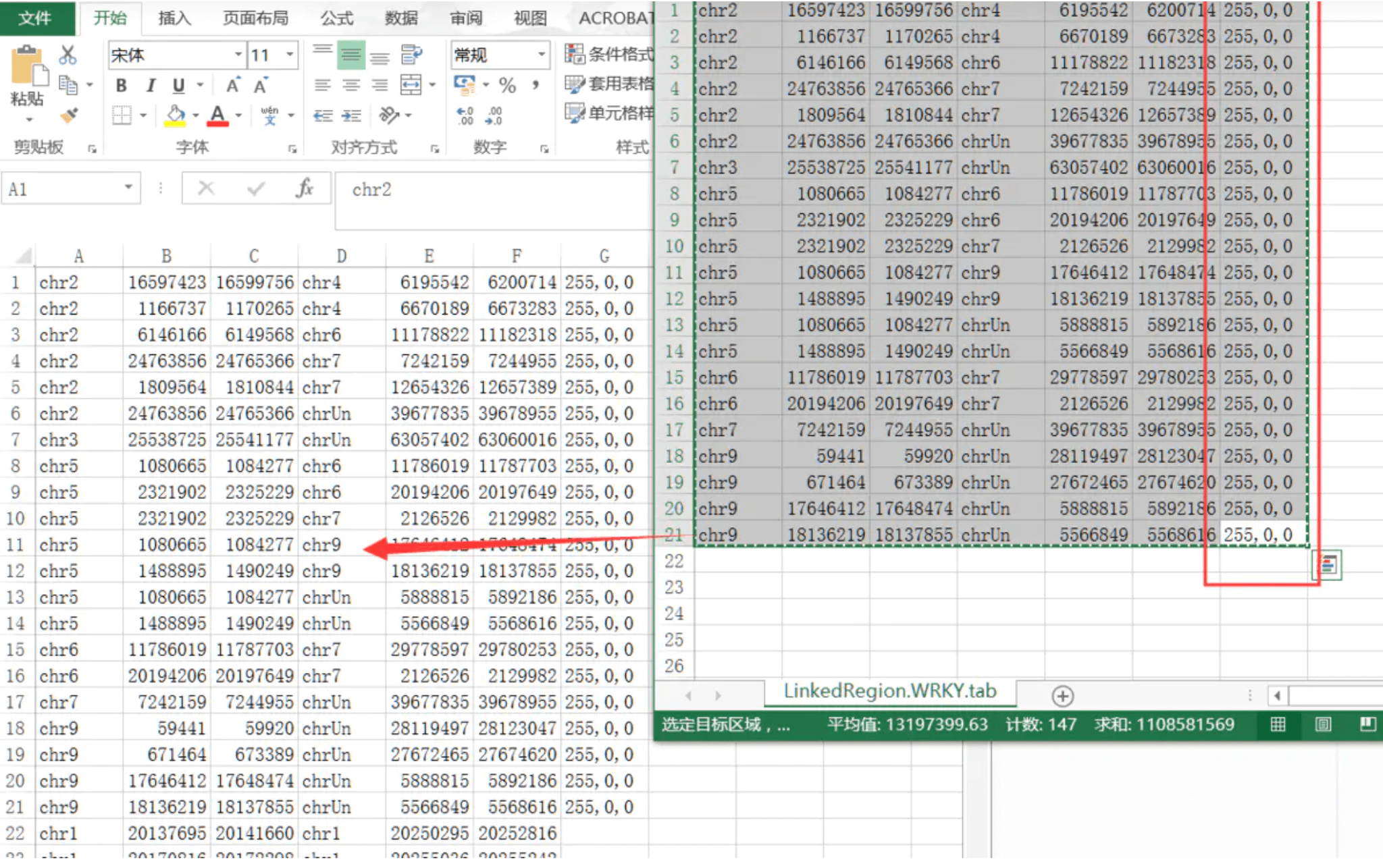
结果图
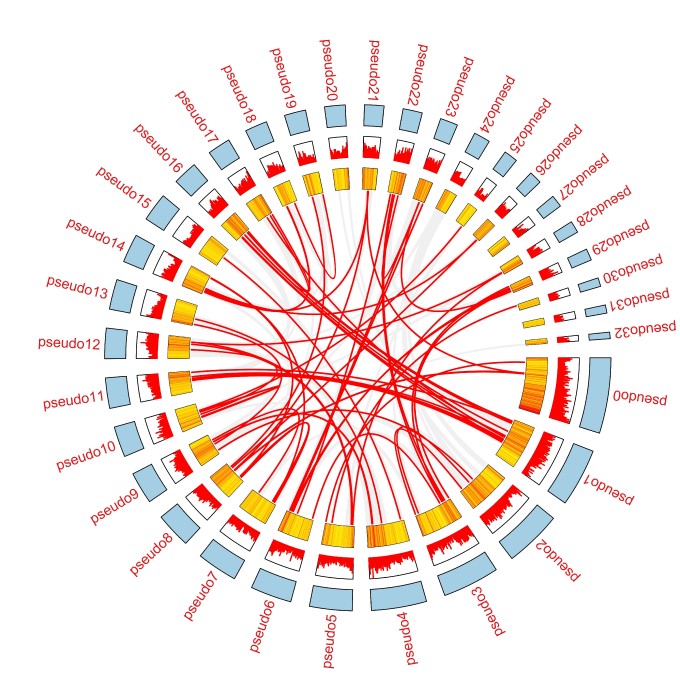
参考
关于TBtools这一部分内容可以参考此处更为详细
点击返回主页 微信
微信 支付宝
支付宝











