
Python 学习教程总结
9c0e01c098a0dd49c427b167e18fce17e67e26ad0a7cba68582557c7576ff6d963a4dae9a8525318485df615608054825dbf0ede2f78eb0f591924f7a5ef0da6f74504f9c5dbf1cb630fefa9a383c55a127917d351aa0b88451d62a05c7ae58a954dbfa69368cd77ff6a998e09a27aaac03e3337a76b80002821b7e1122807d5fbd315b6951fc1a53acdc87ec06e2829bb7861d67bea26d4c92c41e282d7ccb639b07257d1c10addb759cb50b57c1e2d27e37f1fd137857986d03cac9b83fd1af4f975d2c4e7fcb11c3064336f51c7f1ebcf73f8f60166c87c7e5098a0ff2dee57795645c412e5694bdd1c50f941097f096e514571394d4df ...

基因表达数据的聚类分析方法
介绍
基因表达(gene expression) 是指将来自基因的遗传信息合成功能性基因产物的过程。
基因表达产物通常是蛋白质,但是非蛋白质编码基因如转移RNA(tRNA)或小核RNA(snRNA)基因的表达产物是功能性RNA。
所有已知的生命,无论是真核生物(包括多细胞生物)、原核生物(细菌和古细菌)或病毒,都利用基因表达来合成生命的大分子。
基因编码并可用于合成蛋白质,这个过程称为基因表达。
在像人类这样的高等生物中,根据细胞类型(神经细胞或心脏细胞)、环境和疾病状况等各种因素,数以千计的基因以不同的量一起表达。
例如,不同类型的癌症在人类中引起不同的基因表达模式。可以使用微阵列( Microarray )技术研究不同条件下的这些不同基因的表达模式。
微阵列和基因表达谱
来自微阵列的数据可以想象为矩阵或网格,矩阵中的每个单元格对应于特定条件下的基因表达值。
如下图所示,矩阵的每一行对应一个基因 gi ,每一列对应一个条件/样本 si
人类有大约 20,000 个表达基因,假设我们想知道它们的表达模式,即在不同类型的人类癌症下哪些基因产生更高或更低水平的蛋白质。另外,假设已知有 ...
进化树构建之邻接法(Neighbor-Joining)的介绍
进化树构建
进化树构建的问题是推断可能产生给定基因序列数据的进化树的拓扑结构和分支长度。推断树中叶节点的数量应等于给定数据中基因序列的数量。
Neighbor-Joining Algorithm
Neighbor-Joining (NJ)树推理方法最初是由 Saitou 和 Nei 于 1987 年编写的。
它属于一类基于距离的方法用于构建进化树。 NJ 方法采用给定序列之间的成对进化距离矩阵来构建进化树。Neighbor-Joining是一种bottom-up 的聚类方法,常被用于系统发育树 (phylogenetic tree) 的构建当中。
成对距离通常从序列比对算法中获得,例如 Smith-Waterman 和 BLAST ,它们将每个基因序列与每个其他基因序列进行比对。比对得分可用作序列之间进化距离的估计。
可用于计算距离的程序包括:用于 DNA MSA 的 DNADIST 和用于 Protein MSA 的 PROTDIST 。这些程序是 PHYLIP 包的一部分。
我们得到的输出是一棵树以及分支长度
01
使用基于23种遗传信息的邻接法构建的18个人类群体的遗传距离图。 ...

进化树相关概念和基本类型的介绍
介绍
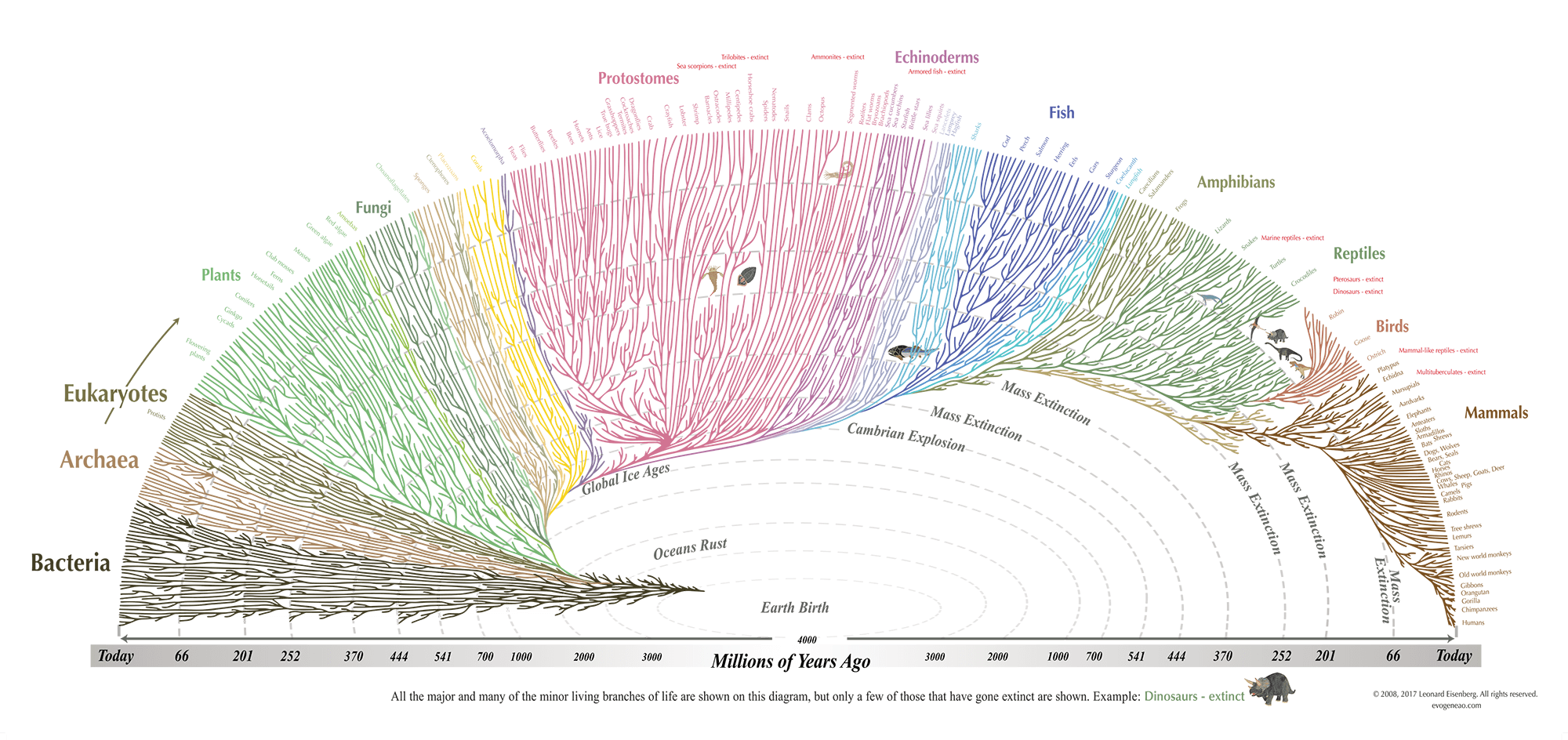
来自形态、生化和基因序列数据的证据表明,地球上的所有生物都具有遗传相关性,生物的谱系关系可以用一棵巨大的进化树、生命之树或进化树来表示。
进化树是一种图,其中正在研究的序列表示为叶节点(leaf nodes),内部节点和分支描述序列之间的进化关系。 在大多数情况下,DNA 序列是来自不同生物体(organisms)的基因序列,可能代表生物体的实际进化。
进化树
分别来自人类、黑猩猩、小鼠和鱼类物种的 4 个基因序列 Human1 、 Chimpanzee1 、 Mouse1 和 Fish1 。
我们还将假设这些是在各自物种中将葡萄糖转化为能量的同源或等效基因(homologous/equivalent genes)。 4个基因的假设进化树可以从下图看出
这棵树显示了来自四个物种的现代或现存基因是如何相互进化的。 树显示有一个共同的祖先基因(树的根)分裂或进化成2个不同的基因; 一个是当今的 Fish1 基因,另一个是小鼠、黑猩猩和人类的共同祖先基因。 然后,小鼠、黑猩猩和人类的共同祖先基因进化成今天的 Mouse1 基因以及 Human1 和 Chimpanzee1 的共同祖先 ...
OrthoFinder 进行直系同源基因分析教程
介绍
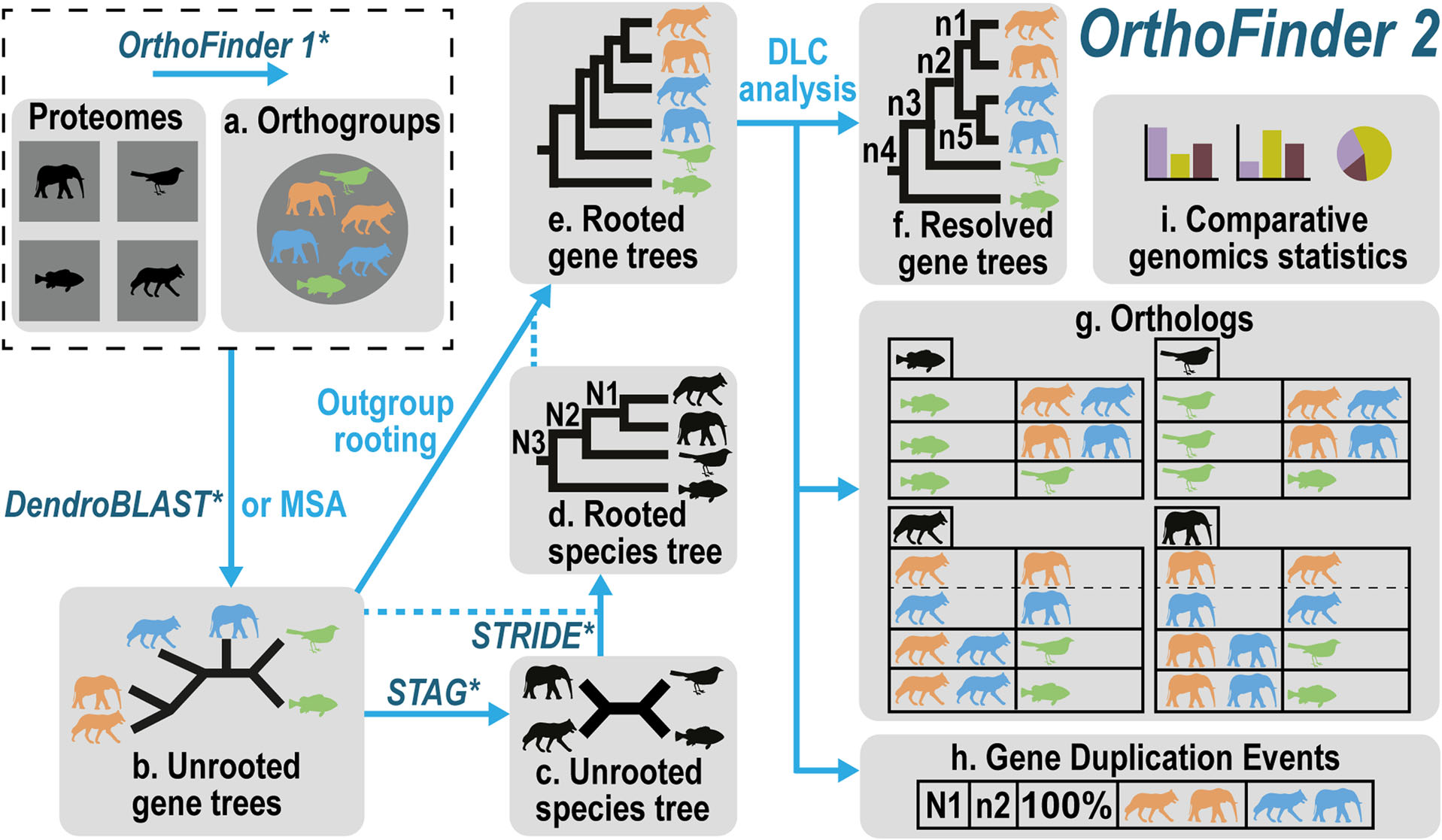
OrthoFinder 是一个快速、准确和全面的比较基因组学平台。 它找到正交群(orthogroups)和直系同源(orthologs),推断所有正交群的有根基因树,并识别这些基因树中的所有基因复制事件。它还为被分析的物种推断出一个有根的物种树,并将基因复制事件从基因树映射到物种树的分支。
OrthoFinder 还为比较基因组分析提供全面的统计数据。 OrthoFinder 使用简单,运行它所需的只是一组 FASTA 格式的蛋白质序列文件(每个物种一个)。
总的来说,它将要分析的物种的蛋白质组作为输入,并从这些蛋白质组中:
推断目标物种的正交群
推断出一组完整的有根基因树
推断有根物种树
使用基因树推断基因之间的所有直系同源关系
推断基因复制事件并将它们交叉引用到基因和物种树上的相应节点
为目标物种提供比较基因组学统计数据
除了大规模分析外,它还可以用于在实验研究之前仔细检查各个直系同源关系。
安装
使用Conda安装
1conda install orthofinder
本地安装
可以使用 Bioconda 安装 OrthoFinder 或直接从 GitHub 下载。 ...
Snakemake 常用参数以及进阶用法介绍
写在前面
上一篇介绍了 Snakemake 入门教程 做了一个简单的示例,具体查看我的上一篇内容
下面会介绍一下 Snakemake的常用参数以及进阶的用法~
介绍之前大家可以看一个视频了解一下(时长:19min14s, 选择性观看)
参数介绍
命令行参数
内核数调用
1234$ snakemake --cores 1# 指定多个可用内核$ snakemake --cores 4
Snakemake 执行在同一目录中 名为 Snakefile 的文件中指定的工作流(Snakefile 可以通过参数 -s 给出)。
试运行
1234$ snakemake -n# 试运行并打印试行内容$ snakemake -n -r
可以进行试运行,测试工作流是否正确定义以及估计所需计算量。
Snakemake 工作流通常定义某些规则的使用线程数。 有时,覆盖工作流定义中给出的默认值可以通过使用--set-threads参数来完成,例如,
1$ snakemake --cores 4 --set-threads myrule=2
将覆盖为rule myrule 定义的 ...
Snakemake 入门教程(创建一个简单的工作流)
写在前面
既然写了教程就需要具有普适性,能适合大多数人的胃口,我这部分的内容以及示例主要还是参考了官方教程,但是都是我一步一步跑过的流程,所以会更有印象,送给想学 Snkaemake 但是一直没有去学的朋友们,这些内容对于有生信基础的人来讲,上手会很快,因为很多的生信软件都使用过,写起来也就没有那么晦涩,下面开始~
Snakemake 定义
Snakemake 工作流管理系统是一种用于创建可重复和可扩展的数据分析的工具。
工作流是通过一种人类可读的、基于 Python 的语言来描述的。它们可以无缝扩展到服务器、集群、grid和云环境,无需修改工作流。
最后,Snakemake 工作流可能需要对所需软件的准备,这些软件将自动部署到任何执行环境。
安装
Snakemake 可在 PyPi 上以及通过 Bioconda 和源代码获得。可以使用任意方法安装 Snakemake,我们这里仅介绍使用 Conda 安装
通过 Conda/Mamba 安装
这是安装 Snakemake的推荐方式,因为Conda安装比较简单。
首先,必须已经安装了一个基于 Conda 的 Python3 发行版。推荐的 ...

学习生物信息学的十条准则
介绍
测序技术正变得比以往任何时候都更加先进和实惠。作为回应,不断壮大的国际联盟,例如地球生物基因组计划 (EBP) 、万种脊椎动物基因组计划 (G10K)、全球无脊椎动物基因组联盟 (GIGA) 、5000种昆虫基因组(i5K)、万种植物基因组计划(10KP),还有许多组织制定了对地球上所有生命进行测序的宏伟计划。
这些联盟旨在利用基因组数据来揭示我们星球生物多样性的生物学秘密,并将这些知识应用于现实世界的问题,例如提高我们对物种进化的理解,协助保护受威胁的物种,以及确定新的医学目标,农业或工业用途。所有这些目标都依赖于有人来分析和理解海量的生物数据,这使得bioinformaticians比以往任何时候都更加抢手。许多具有生物学和遗传学背景的研究人员正在加紧迎接大数据分析的挑战,但是如果没有强大的计算和/或计算机科学。最近的一篇“十大简单规则”文章强调了生物信息学研究支持的重要性。
在这里,为有兴趣进入生物信息学领域以及那些开始生物信息学之旅的人提供 10 条简单规则。无论你是学生、经验丰富的生物学家或遗传学家,还是可能对这一新兴领域感兴趣的任何人。这些规则用于开始使用命令行学 ...

rust-mdbg 一款用于基因组组装的高效率软件
写在前面
rust-mdbg 是一种超快的minimizer-space de Bruijn graphs (mdBG) 实现,适用于组装长而准确的读数,例如PacBio HiFi。
随着18年以来Pacbio HiFi reads的出现,让一些复杂基因组的组装不再复杂,而且有越来越多的课题组也加入到了基因组学的研究中,正是因为有了高精度长读长的reads,目前也产生了很多专门用于HiFi组装的软件,如Hifiasm,当然这篇文章的软件的算法,可以用超短时间,低内存去组装。我相信随着不断的发展,以后做组装的时候甚至都不需要服务器,在个人电脑也可以实现。
内容写了很多,考虑到阅读体验,做了删减。
下面是正文~
原理介绍
DNA测序技术发展的很快,尤其是以Pacbio HiFi数据为代表的Long reads兼顾了长读长以及高准确度。
在这里,研究者定义了一种算法方法mdBG,它利用最小空间德布莱英图(de Bruijn graph)实现long reads基因组组装。
这里插入一个视频,让大家了解一下以前的组装软件用到的 de Bruijn graph的原理
...

Perl语言基础(一)
简介
什么是Perl语言?
Perl一般被称为“实用报表提取语言”( Practical Extraction and Report Language)
Perl最初的设计者为拉里沃尔(Larry Wall)他于1987年12月18日发表。
“Perl”,有大写的P,是指语言本身;"perl”,小写的p,是指程序运行的解释器。
Perl语言特点
1、开源免费
2、支持跨平台
3、直接运行,无需复杂编译
4、内部集成正则表达式
5、CPAN模块库
为什么Perl语言适合处理生物数据
1、正则表达是善于处理字符串
2、脚本语言,善于批量化和流程化
3、很多生物学软件用Perl语言编写
4、简单易学
基础语法
Perl 程序有声明与语句组成,程序自上而下执行,包含了循环,条件控制,每个语句以分号 (;) 结束。
Perl 语言没有严格的格式规范,你可以根据自己喜欢的风格来缩进。
单引号和双引号
12345#!/usr/bin/perl print "Hello, world\n"; # 双引号print 'Hello, world\n'; # 单引号
输出结果如下:
H ...
公告
如果你有更多想了解的内容,欢迎关注我的个人公众号:生信技术





