
使用 RGL 制作交互式 3D 散点图
介绍
本 R 教程 逐步描述了如何使用 rgl包 构建 3D 图形。
RGL 是一个 3D 图形包,可生成实时交互式 3D 绘图。
它允许交互式旋转、缩放图形和选择区域。
rgl 包还包括一个名为 R3D 的通用 3D 接口。
R3D 是本文末尾描述的通用 3D 对象和函数的集合。
内容14913字 32图!
安装 RGL 包
1install.packages("rgl")
在 Linux 操作系统上,可以按如下方式安装rgl包:
1sudo apt-get install r-cran-rgl
加载 RGL 包
1library("rgl")
准备数据
我们将在以下示例中使用iris数据集:
123456data(iris)head(iris)x <- sep.l <- iris$Sepal.Lengthy <- pet.l <- iris$Petal.Lengthz <- sep.w <- iris$Sepal.Width
启动和关闭 RGL device
要使用 RGL 制作 3D 绘图,应该首先在 R 中启动 RGL device
以 ...
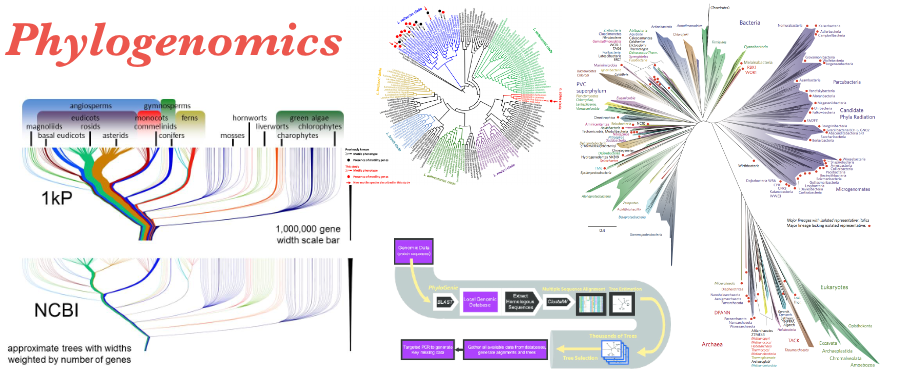
系统发育基因组学(Phylogenomics)的介绍以及实操
Phylogenomics
写在前面
关于系统发育基因组学的内容,本文参考了 Mike Lee 的文章,将分为两期,本文介绍基本相关原理。
有一个相关的视频,时长为32’51’’ 感兴趣的可以点击下方进入观看。
什么是系统基因组学?
用一个容易理解但是不准确的概念来表示: 系统基因组学正试图在基因组水平上推断进化关系。
因为在实践中,我们并没有关注的所有生物体整个基因组。
并且根据我们考虑的多样性的广度,无论如何使用整个基因组是不可能或毫无意义的(因为它们可能太不同了)。
所以更恰当的说法是:系统基因组学试图在更接近基因组的水平上推断进化关系,而不是单个基因的系统发育(比如16S rRNA 基因树)。
一般大家习惯于查看和使用的大多数系统发育树是单一基因类型(如 16S rRNA 基因)的不同拷贝去估计进化关系的视觉表示。
如果我们试图在生物体层面上思考,我们将使用这些基因拷贝作为代表生物体本身的代理,并且我们假设这些基因的进化关系告诉我们一些关于它们源生物进化关系的有意义的事情。
我们正在用系统基因组学做同样的事情,只是我们使用多个基因而不是单个基因。
...
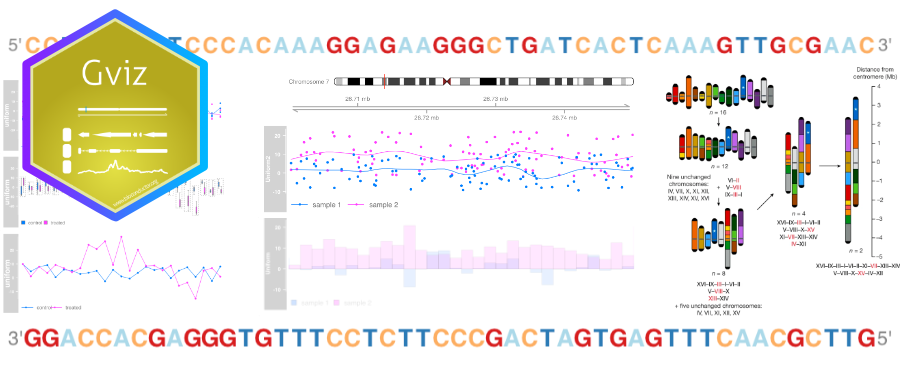
Gviz - 实现基因组数据可视化
Gviz包介绍
Gviz软件包简介
Gviz软件包旨在提供一个结构化的可视化框架,以沿着基因组坐标绘制任何类型的数据。它还允许整合来自UCSC或ENSEMBL等来源的公开基因组注释数据。
与大多数基因组浏览器一样,单独类型的基因组特征或数据由单独的Track表示。
默认情况下,Gviz 检查所有提供的染色体名称在 UCSC 上的有效性(染色体必须以 chr 字符串开头)。可以决定通过调用 options(ucscChromosomeNames=FALSE) 来关此功能
在以下示例中,将利用小鼠 mm9 基因组上的 UCSC 基因组和 7 号染色体 (chr7)
绘制 AnnotationTrack 图
请注意,AnnotationTrack 构造函数可以容纳许多不同类型的输入。
例如,注释功能的开始和结束坐标可以作为单个参数
start和end作为 data.frame 或甚至作为 IRanges 或 GRangesList 对象。
123456789101112131415161718192021library(Gviz)library(GenomicRanges)#加载数据 : ...

R数据可视化|使用Scatterplot3d包制作3D散点图
介绍
R 中有许多包(RGL、car、lattice、scatterplot3d等)用于创建3D 图形。
本教程介绍了如何使用 R 的 scatterplot3d包 在 3D 空间中生成散点图。
scaterplot3d 使用起来非常简单,可以通过在已经生成的图形中添加补充点或回归平面来轻松扩展。
它可以很容易地安装,因为它只需要一个已安装的 R 版本。
安装并加载 scaterplot3d
12install.packages("scatterplot3d")library("scatterplot3d")
准备数据
iris 数据集将被使用进行画图:
12data(iris)head(iris)
Iris 也称鸢尾花卉数据集,是一类多重变量分析的数据集。数据集包含150个数据样本,分为3类,每类50个数据,每个数据包含4个属性。可通过花萼长度,花萼宽度,花瓣长度,花瓣宽度4个属性预测鸢尾花卉属于(Setosa,Versicolour,Virginica)三个种类中的哪一类。
函数 scatterplot3d()
一个简化的格式是:
12scatterplot3d(x, y ...

使用最长公共子序列算法进行序列比对
介绍
在分子生物学中,DNA 和蛋白质可以表示为字母序列。
DNA 序列由 A、T、G、C 组成,代表核苷碱基(nucleobases) 腺嘌呤、胸腺嘧啶、鸟嘌呤和胞嘧啶。
蛋白质由 20 个不同的字母组成,表示 20 种不同的氨基酸。
比较来自同一生物体或来自不同生物体的两个序列,称为 序列比较 (Sequence comparison),是分子生物学中的一项重要任务。
它有助于为许多生物学问题提供解决方案,例如:
预测蛋白质的结构和功能
推断物种的进化历史和相关性
定位基因/蛋白质中的常见子序列以识别常见基序,
作为 DNA 测序的基因组组装中的一个子问题
为了进行序列比较,我们首先进行 序列比对(sequence alignment)。
什么是序列比对?
序列比对是将一个序列置于另一个序列之上以识别相似字符或子串之间的对应关系的一种方式。
它可以在脱氧核糖核酸 (DNA)、核糖核酸 (RNA) 或蛋白质序列上进行。
来自不同生物体的序列可能具有不同的大小。对齐需要在序列的任意位置插入空格,以便两者具有相同的大小。
在序列的开头或结尾插入“空格”或“间隙”。
让我们考虑一个例子 ...

PyPubMed|一款进行文献检索的工具
写在前面
其实这个工具在年初的时候使用过,最近翻看自己以前的笔记时候又看到了。
我觉得这款工具是很不错的文献检索工具,具体的操作以及参数也比较简单。
搜索指定关键词就可以查到相关的文献名称、摘要(CN/EN)以及DOI号等信息。
对于了解相关领域/关键词的文献还是很有帮助。
工具开发作者:苏庆东
安装 PyPubMed
12345678910# 要求Python环境 Python3.6+ pip3 isntall pypubmed#若安装速度太慢报错,可使用镜像来加速,输入下面命令:# 清华镜像pip3 install pypubmed -i https://pypi.tuna.tsinghua.edu.cn/simple# 豆瓣镜像pip3 install pypubmed -i https://pypi.douban.com/simple# 阿里云镜像pip3 install pypubmed -i https://mirrors.aliyun.com/pypi/simple
安装后,测试一下安装是否成功,输入下方命令行:
pypubmed
出现如下提示,表示安装成功:
1234 ...
Python 进阶笔记
9c0e01c098a0dd49c427b167e18fce17e67e26ad0a7cba68582557c7576ff6d979a89f18e7df0a66953726a25b6d80b8e16018c3cbde2b206323563c29ed6852b16307d4fd5722ee5610ac2bff07581ec16669bdb29675830e5afb6385ffc9b06ba54cd6f7af45188fc19e5f63fa2b6c64a454cda7a448504987ed3afa934a5d8fc62a1d2dd7e4fe89ee5ae90fc6445afe54a578cd35c0726254af56c1b1448aec7e8fb4fa5f7f1bd0ee7acf6f0d1d1ea6bf1272f12f3ef86c5a3efa9dbf07841367ce456fb83c8803257851d2c891f5f6fc3b5ca4368f8aef8a810237767e7dfcfef54b65117efeb309f6a5572c3c31525fa404629890c6b ...

保守序列的生物学意义|用 K-mers 寻找保守序列
在这篇文章中,我们将介绍保守序列的概念,并描述它们的生物学意义。
然后,我们将看到如何将寻找保守序列的问题简化为在给定序列中找到最常见 K-mer 的问题,并进一步修改该问题以处理不匹配,从而使我们的问题在生物学上更合理。
最后,我们将看到一个简单的算法来解决不匹配 K-mer 的问题。
保守序列
在进化生物学和遗传学中,保守序列是指代代发生在不同或相同物种中的相同或相似的 DNA 或 RNA 或氨基酸(蛋白质)序列。这些序列的组成变化很小,有时甚至几代都没有变化。
以下示例显示了跨物种的序列保守实际上是什么样的:
在这张图片中,我们可以看到哺乳动物组蛋白的氨基酸序列及其保守区域。
灰色表示的那些在所有物种中都是保守的,白色的差距往往随着不同物种的变化而变化。
保守序列的常见例子
在基因组中多处发现保守的翻译和转录相关序列
发现核糖体中的某些 RNA 成分在不同物种中高度保守
发现 tmRNA 在多种细菌物种中是保守的
其他例子,如 TATA(重复区域)和 同源异型盒 (homeoboxes)(参与调节多种物种的胚胎发育)。
NCBI 提供的保守域数据库拥有关于不 ...

R语言基础画图教程合集(长文总结)
前言
这一部分会简单分享一些R语言画图的小技巧,后续相应的代码以及测试数据文件都已经上传到百度网盘,可以在**公众号(生信技术)**留言回复“ R语言 ”获取。
R包安装
需要安装 ggplot2, qqman, gplots, pheatmap, scales, reshape2, RColorBrewer 和 plotrix(使用 install.packages(), 如 install.packages('ggplot2')
下面是后续需要用到的一些R包,如果需要新下载后面文章会继续介绍。
12345678910library(ggplot2)library(qqman)library(gplots)library(pheatmap)library(scales)library(reshape2)library(RColorBrewer)library(plyr)library(plotrix)library(ggpubr) #用于带统计检验的箱线图
加载数据
1234567891011121314151617181920212223# Read the input files ...
使用同源建模预测蛋白质结构
什么是蛋白质?
蛋白质是大的生物分子,负责执行生物体细胞内的大部分功能,包括对刺激作出反应、作为其他反应的催化剂、将分子从一个地方运输到另一个地方以及执行细胞信号传导。就像 DNA 序列一样,蛋白质序列是一串分子,但与 DNA 序列不同的是,有20种不同的称为氨基酸的分子构成了蛋白质序列。
蛋白质结构
每个1D 蛋白质序列串都折叠成3D 结构。这些 3D 蛋白质结构决定了蛋白质如何响应各种环境以及它与哪些其他分子相互作用,因此对于蛋白质执行其功能的能力至关重要。蛋白质的 3D 结构是通过提供蛋白质中每个原子在3D 空间中的坐标 (xyz)来描述的。
确定蛋白质结构
可以使用X 射线晶体学和核磁共振 (NMR)等实验程序确定蛋白质结构。然而,这些技术缓慢且繁琐,并且不能应用于所有蛋白质。因此,高通量计算方法用于从序列预测蛋白质的 3D 结构。
同源建模
蛋白质结构预测最流行的计算方法之一是同源建模。同源建模利用蛋白质结构的进化保守性来预测蛋白质的 3D 结构。从相同的共同祖先(同源性)进化而来的两种蛋白质往往具有相似的 3D 结构。
在同源建模中,这种蛋白质结构保守性的特性用于预测新发 ...
公告
如果你有更多想了解的内容,欢迎关注我的个人公众号:生信技术





